Die kommerzielle Nutzung dieses Inhalts ist strengstens untersagt. Weitere Details zur Lizenzrichtlinie finden Sie auf der Seite Über uns.
Teil I. Makroskopische Thermodynamik
1.1 Gleichgewichtszustand und Zustandsgrößen
1.1.1 Gleichgewichtszustand
Thermodynamisches Gleichgewicht stellt einen fundamentalen Zustand dar, in dem die makroskopischen Eigenschaften eines Systems über die Zeit ohne äußere Einflüsse konstant bleiben. Dieses statische Erscheinungsbild verbirgt eine intensive mikroskopische Aktivität – Moleküle bewegen sich und stoßen weiter zusammen, aber ihr kollektives Verhalten erzeugt stabile makroskopische Mittelwerte. Eine kritische Unterscheidung besteht zwischen echtem Gleichgewicht und stationären Zuständen: Während beide konstante Eigenschaften aufweisen, erfordern stationäre Zustände einen kontinuierlichen Energie-/Materieaustausch mit der Umgebung, wie z. B. ein Metallstab, der einen Temperaturgradienten zwischen 0 °C und 100 °C an den Enden aufrechterhält. Echtes Gleichgewicht erfordert drei gleichzeitige Bedingungen: thermische Gleichmäßigkeit (keine Temperaturgradienten), mechanische Gleichmäßigkeit (keine Druckgradienten) und chemische Gleichmäßigkeit (keine Zusammensetzungsgradienten).
Zustandsgrößen quantifizieren diese Gleichgewichtseigenschaften. Das Volumen repräsentiert den verfügbaren Raum für die Molekülbewegung, definiert als das Innenvolumen des Behälters abzüglich des ausgeschlossenen Volumens der Moleküle selbst. Der Druck ist die senkrechte Kraft pro Flächeneinheit, die von Molekülen auf die Behälterwände ausgeübt wird, und entsteht durch Impulsübertragung bei Kollisionen. Für Systeme ohne elektromagnetische Effekte oder chemische Reaktionen genügen und allein zur Definition des Zustands – solche Systeme werden als einfache -Systeme bezeichnet. Das Konzept des Gleichgewichts ist idealisiert, aber essentiell, da reale Systeme durch Teilchenwechselwirkungen ständig auf diesen Zustand hin evolvieren.
1.1.2 Temperatur
Das Nullte Gesetz der Thermodynamik liefert die rigorose Grundlage für die Existenz der Temperatur. Betrachten Sie drei Systeme A, B und C. Wenn A und C thermisches Gleichgewicht erreichen, erfüllen ihre Zustandsgrößen:
Ähnlich ergibt das B-C-Gleichgewicht:
Das Nullte Gesetz behauptet, dass dann auch das A-B-Gleichgewicht gelten muss. Mathematische Analyse zeigt, dass sich diese Beziehungen in eine universelle Funktion faktorisieren lassen:
Gleichsetzen der Ausdrücke beweist, dass gilt. Diese universelle Funktion definiert die Temperatur – eine intensive Größe, die für Systeme im thermischen Gleichgewicht identisch ist.
Temperaturskalen implementieren dieses Konzept. Empirische Skalen wie Fahrenheit () und Celsius entstanden aus der praktischen Thermometrie, leiden aber unter materialabhängiger Nichtlinearität. Die ideale Gastemperaturskala löst dieses Problem, indem sie Gase bei geringer Dichte nutzt, wo intermolekulare Kräfte vernachlässigbar werden. Für ein Gasthermometer bei konstantem Volumen:
wobei der Druck am Tripelpunkt von Wasser (273,16 K) ist. Dieser Grenzwertprozess eliminiert gasespezifische Verhaltensweisen und gewährleistet eine universelle Temperaturdefinition. Die thermodynamische (Kelvin-)Skala ist fundamental durch die Carnot-Effizienz definiert:
für reversible Wärmekraftmaschinen zwischen Reservoirs bei und , die, wo anwendbar, mit der idealen Gastemperaturskala übereinstimmt.
Moderne praktische Skalen wie ITS-90 implementieren thermodynamische Temperaturen unter Verwendung spezifizierter Fixpunkte und Instrumente: Gastheorie unter 24 K, Platinwiderstandsthermometer (13,8 K–961 °C) und Strahlungsthermometrie über 961 °C. Die Kelvin- (K) und Celsius-Skala (°C) sind durch verbunden und behalten identische Einheitsgrößen für Temperaturintervalle bei.
1.1.3 Zustandsgleichung
Die Zustandsgleichung stellt fundamentale Beziehungen zwischen thermodynamischen Parametern im Gleichgewicht her. Drei experimentelle Gasgesetze bilden die Grundlage:
Boyle-Mariottesches Gesetz ( bei konstanter ) etabliert die isotherme Kompressibilität
Gay-Lussac-Gesetz ( bei konstantem ) definiert den volumetrischen Ausdehnungskoeffizienten
Charles-Gesetz ( bei konstantem ) liefert den Druckkoeffizienten
Die ideale Gasgleichung synthetisiert diese Beobachtungen mit dem Avogadro-Prinzip. Ausgehend vom Boyle-Mariotte-Gesetz betrachten wir einen Prozess bei konstantem Druck mit dem Tripelpunkt als Referenz:
In Kombination mit dem Boyle-Mariotte-Gesetz am Tripelpunkt ergibt sich:
Das Avogadro-Gesetz bestätigt, dass für alle Gase bei gegebener Stoffmenge identisch ist und die universelle Konstante definiert:
wobei das molare Volumen am Tripelpunkt ist.
Mikroskopisch verbindet die Boltzmann-Konstante die makroskopische und molekulare Beschreibung:
Für Gasgemische kombiniert sich Daltons Gesetz () mit Partialdrücken zu:
was die Zustandsgleichung des Gemisches beweist.
Reale Gase weichen bei hohen Dichten und niedrigen Temperaturen von der Idealität ab. Die van-der-Waals-Gleichung berücksichtigt das Molekülvolumen () und die Anziehung ():
Kritische Parameter ergeben sich aus mathematischen Bedingungen am kritischen Punkt:
Die simultane Lösung dieser Gleichungen liefert:
Der reduzierte Kompressibilitätsfaktor liefert einen universellen Bezugspunkt. Für breitere Genauigkeit korrigiert die Onnes-Virialentwicklung:
korrigiert systematisch Abweichungen durch temperaturabhängige Virialkoeffizienten, die mittels Gas isothermalkompression messbar sind.
1.2 Mikroskopisches Modell der Materie
1.2.1 Einführung
Die Grundlage der kinetischen Gastheorie liegt im Verständnis der Materie auf ihrer fundamentalsten Ebene. Alle makroskopischen Substanzen – ob Festkörper, Flüssigkeiten oder Gase – bestehen aus einer großen Anzahl mikroskopischer Teilchen, genannt Atome oder Moleküle, die durch leeren Raum getrennt sind. Diese atomare Struktur erklärt, warum Materialien komprimiert werden können: Die scheinbare Festigkeit der Materie ist eine Illusion, die durch elektromagnetische Kräfte zwischen den Teilchen entsteht, nicht durch tatsächlichen Kontakt zwischen ihnen. Moderne wissenschaftliche Instrumente wie Rastertunnelmikroskope haben diese unsichtbare Welt sichtbar gemacht und uns erlaubt, einzelne Atome abzubilden und sie sogar zu Strukturen wie Buchstaben oder Mustern zu manipulieren.
Die unaufhörliche, chaotische Bewegung dieser Teilchen bildet das Herzstück thermischer Phänomene. Diese Bewegung intensiviert sich mit der Temperatur, wie eindrucksvoll durch die Brownsche Bewegung demonstriert – den zufälligen Tanz von Pollenkörnern oder Rauchpartikeln, die in einer Flüssigkeit suspendiert sind. Unter dem Mikroskop beobachtet, zittert diese Partikel unvorhersehbar aufgrund unausgeglichener Kollisionen mit den umgebenden Flüssigkeitsmolekülen. Je kleiner das Partikel, desto heftiger wird seine Bewegung, was direkten Beweis dafür liefert, dass das, was wir als "ruhige" Flüssigkeit oder Gas wahrnehmen, tatsächlich ein Rauschzustand molekularer Aktivität ist. Diese ständige Bewegung ist nicht auf Flüssigkeiten beschränkt; selbst in Festkörpern schwingen Atome um feste Positionen wie Federn, die ein molekulares Gerüst verbinden.
Wechselwirkungen zwischen Molekülen steuern die Materialzustände durch konkurrierende Kräfte. In extrem geringer Entfernung (< 0,1 nm) dominieren starke Abstoßungskräfte, wenn sich Elektronenwolken überlappen, was verhindert, dass Materie zu unendlicher Dichte kollabiert. In mittleren Entfernungen (0,1–1 nm) übernehmen Anziehungskräfte durch elektromagnetische Wechselwirkungen zwischen temporären Dipolen in ansonsten neutralen Molekülen – die van-der-Waals-Kräfte, die Flüssigkeiten Kohäsion verleihen. Diese gegensätzlichen Kräfte erzeugen einen Gleichgewichtsabstand, bei dem sich Moleküle natürlich einpendeln, wie Tänzer, die in einem überfüllten Raum persönlichen Raum wahren. Das empfindliche Gleichgewicht zwischen molekularer Bewegung und diesen Wechselwirkungskräften erklärt Phasenübergänge: Erhitzen liefert kinetische Energie, um die Anziehung zu überwinden, und wandelt Feststoffe in Flüssigkeiten und dann in Gase um.
1.2.2 Druck idealer Gase
Um den Gasdruck auf molekularer Ebene zu verstehen, konstruieren wir ein vereinfachtes Modell, das wesentliche Verhaltensweisen erfasst und komplexe Details ignoriert. Stellen Sie sich Gasmoleküle als infinitesimale Punkte anstelle von physischen Objekten vor – eine vernünftige Annäherung, da Moleküldurchmesser (~10⁻¹⁰ m) von typischen intermolekularen Abständen (~10⁻⁹ m bei STP) dwarfed werden. Zwischen Kollisionen bewegen sich diese Teilchen frei, ohne gegenseitige Anziehung oder Abstoßung, wie Pendler, die sich in einem riesigen Bahnhof ignorieren. Kollisionen zwischen Molekülen oder mit Behälterwänden erfolgen augenblicklich und elastisch, wobei sowohl Impuls als auch kinetische Energie wie perfekte Billardkugelstöße erhalten bleiben.
Diese Idealisierung ergibt sich natürlich aus experimentellen Beobachtungen: Gase dehnen sich aus, um Behälter zu füllen, weil sich Moleküle unabhängig bewegen; geringe Dichten erleichtern die Kompression durch Verringerung der intermolekularen Abstände; konstanter Druck im Gleichgewicht impliziert gleichmäßige Kollisionsraten. Die Stärke des Modells liegt darin, die chaotische Komplexität von 10²³ Molekülen in handhabbare Statistiken zu verwandeln, bei denen individuelle Bahnen irrelevant werden und kollektive Mittelwerte dominieren.
1.2.3 Statistische Annahmen im Gleichgewicht
Wenn ein Gas das thermodynamische Gleichgewicht erreicht, entstehen trotz fortwährendem molekularem Chaos zwei mächtige statistische Prinzipien. Erstens verteilen sich die Moleküle gleichmäßig im Raum – jedes makroskopische Volumenelement enthält ungefähr die gleiche Teilchenzahl, unabhängig vom Ort. Diese räumliche Homogenität erlaubt die Definition der Teilchendichte als Konstante im gesamten Behälter. Zweitens zeigen die Molekülgeschwindigkeiten keine gerichtete Präferenz; alle drei kartesischen Komponenten teilen identische statistische Eigenschaften:
Die erste Reihe zeigt keinen Nettostrom an, während die zweite gleiche Verteilung der kinetischen Energie über die Dimensionen hinweg offenbart. Diese Symmetrien verwandeln das unlösbare Problem der Verfolgung einzelner Moleküle in handhabbare statistische Mechanik.
1.2.4 Formel für den Druck idealer Gase
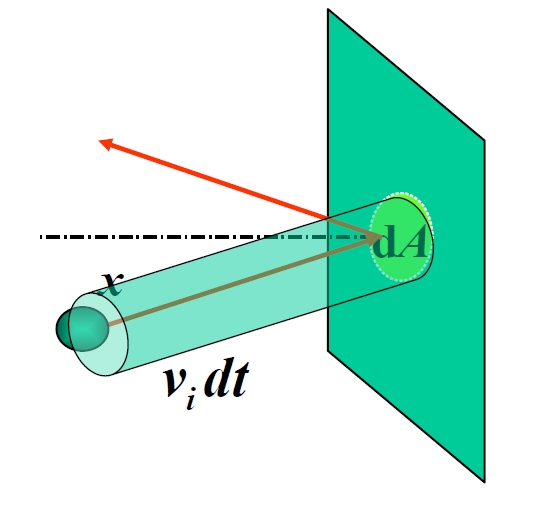
Der Druck entsteht aus dem unaufhörlichen Anprall molekularer Stöße auf die Behälterwände. Betrachten Sie Moleküle, die sich einer Wandfläche senkrecht zur x-Achse nähern. Jeder Stoß kehrt die x-Komponente des Impulses um und liefert den Impuls an die Wand. Um die Gesamtkraft zu ermitteln, berechnen wir, wie viele Moleküle in der Zeit treffen. Moleküle mit der Geschwindigkeit innerhalb der Entfernung können die Wand erreichen und bilden einen imaginären Stoßzylinder mit dem Volumen . Bei einer Teilchendichte für die Geschwindigkeitsgruppe ist die Anzahl der Stöße .
Summieren der Impulse aller Geschwindigkeitsgruppen:
Unter Verwendung der Geschwindigkeitsisotropie und der Definition der translatorischen kinetischen Energie :
Diese elegante Ableitung zeigt den Druck als statistische Manifestation der molekularen kinetischen Energie.
Diagramm des Drucks auf die Gefäßwand
1.2.5 Mikroskopische Interpretation der Temperatur
Die Verbindung zwischen mikroskopischer Bewegung und Temperatur beginnt mit der Kombination der Druckgleichung mit dem makroskopischen idealen Gasgesetz . Gleichsetzen dieser ergibt die tiefgreifende Beziehung:
Diese einfache Formel hat tiefgreifende Auswirkungen. Temperatur misst direkt die durchschnittliche kinetische Energie der molekularen Translation – eine universelle Währung, bei der alle Gasmoleküle unabhängig von ihrer Masse den gleichen Wert haben. Bei Raumtemperatur (300 K) trägt jedes Molekül – vom leichten Wasserstoff bis zum schweren Xenon – etwa J translatorische Energie. Diese Energieäquipartition erklärt, warum leichtere Moleküle sich schneller bewegen, um die gleiche Energie zu erreichen:
Zum Beispiel rasen Wasserstoffmoleküle ( kg) bei 300 K mit etwa 1920 m/s, während Sauerstoffmoleküle ( kg) mit gemächlicheren 480 m/s unterwegs sind.
Die kollektive Natur der Temperatur wird bei Betrachtung von Gasgemischen deutlich. Daltons Gesetz ergibt sich natürlich, da alle Komponenten im Gleichgewicht die gleiche teilen. Diese Energiegleichheit besteht auch zwischen unähnlichen Molekülen, da Kollisionen kinetische Energie umverteilen, bis ein Gleichgewicht erreicht ist – ähnlich wie Billardkugeln unterschiedlicher Masse nach zahlreichen Stößen die gleiche durchschnittliche Energie erreichen.
1.2.6 Molekulare Kräfte
Unter der scheinbaren Einfachheit von Gasen verbirgt sich ein komplexes Zusammenspiel elektromagnetischer Kräfte. Jedes Molekül enthält positiv geladene Kerne, umgeben von negativ geladenen Elektronen. Wenn sich Moleküle nähern, verzerren sich ihre Elektronenwolken, wodurch temporäre Dipole entstehen, die Anziehungskräfte erzeugen – die van-der-Waals-Wechselwirkung. In geringeren Abständen dominiert die Pauli-Abstoßung, wenn überlappende Elektronenorbitale einer Kompression widerstehen.
Diese konkurrierenden Effekte erzeugen die charakteristische molekulare Kraftkurve: stark abstoßend unterhalb des Gleichgewichtsabstands , anziehend oberhalb von und vernachlässigbar jenseits mehrerer Nanometer. Physiker modellieren dieses Verhalten mit Potenzialenergiefunktionen:
Lennard-Jones-Potenzial:
balanciert kurzreichweitige Abstoßung (r⁻¹²-Term) mit längerreichweitiger Anziehung (r⁻⁶-Term). Das Minimum bei repräsentiert den optimalen Bindungsabstand.
Sutherland-Potenzial:
modelliert undurchdringliche harte Kugeln mit schwacher Anziehung.
Hartkugel-Potenzial:
ignoriert die Anziehung vollständig und konzentriert sich auf ausgeschlossene Volumeneffekte.
Diese Modelle dienen unterschiedlichen Zwecken: Lennard-Jones beschreibt Edelgase genau, Sutherland vereinfacht die van-der-Waals-Theorie, während Hartkugel-Modelle helfen, dichte Flüssigkeiten zu verstehen.
1.2.7 Druck in van-der-Waals-Gasen
Reale Gase weichen aufgrund zweier molekularer Effekte vom idealen Verhalten ab: endliche Größe und gegenseitige Anziehung. Johannes van der Waals modifizierte genial das ideale Gasgesetz, um beide zu berücksichtigen. Erstens nehmen Moleküle physischen Raum ein, was das verfügbare Volumen für die Bewegung reduziert. Für Moleküle ist das ausgeschlossene Volumen nicht einfach mal das Molekülvolumen, da der Ausschluss paarweise Wechselwirkungen beinhaltet. Statistische Analyse zeigt:
wobei der effektive Moleküldurchmesser ist. Diese Korrektur transformiert das Volumen im idealen Gasgesetz zu .
Zweitens reduzieren Anziehungskräfte den Druck. Oberflächenmoleküle in der Nähe von Behälterwänden erfahren Nettokräfte nach innen von den Molekülen im Volumen, was ihre Stöße schwächt. Dieser "innere Druck" skaliert mit dem Produkt der Dichten der anziehenden und angezogenen Moleküle:
Unter Verwendung des Sutherland-Potenzials kann die Konstante aus der Potenzialtiefe und dem Bereich abgeleitet werden:
Die Kombination beider Korrekturen ergibt die van-der-Waals-Gleichung:
Dies erklärt elegant das Verhalten realer Gase wie Verflüssigung und kritische Phänomene.
1.3 Verteilung der Molekülgeschwindigkeiten und Energie
Die Verteilung der Molekülgeschwindigkeiten in einem idealen Gas im thermischen Gleichgewicht wird aus fundamentalen statistischen Prinzipien abgeleitet. Betrachten Sie die Geschwindigkeitsverteilungsfunktion , definiert so, dass die Wahrscheinlichkeit, dass ein Molekül Geschwindigkeitskomponenten zwischen und , und und und hat, ist. Für ein isotropes System hängt diese Funktion nur von der Geschwindigkeit ab, und die Geschwindigkeitskomponenten sind statistisch unabhängig. Diese Annahmen führen zur funktionalen Form . Die Logarithmierung und Anwendung der Variablentrennung ergibt , was zu einer Gaußschen Verteilung führt:
Die Normierungsbedingung bestimmt durch Gaußsche Integrale. Der Äquipartitionssatz fixiert und ergibt die Maxwellsche Geschwindigkeitsverteilung:
Um die Geschwindigkeitsverteilung zu erhalten, integrieren wir über alle Geschwindigkeitsrichtungen (Kugelkoordinaten):
Somit beschreibt die Wahrscheinlichkeitsdichte für Molekülgeschwindigkeiten.
1.3.2 Charakteristische Geschwindigkeiten und Verteilungseigenschaften
Die wahrscheinlichste Geschwindigkeit tritt am Maximum von auf. Die Lösung von ergibt:
Die Durchschnittsgeschwindigkeit wird durch Integration von gewichtet mit berechnet:
Unter Verwendung der Substitution und Gammafunktionen ergibt sich:
Die quadratische Mittelgeschwindigkeit leitet sich von der mittleren quadratischen Geschwindigkeit ab:
Somit ist . Die Temperaturabhängigkeit entsteht, weil ist, was zu einer Verbreiterung der Verteilung führt. Die Massenabhängigkeit impliziert, dass schwerere Moleküle schmalere Verteilungen aufweisen.
1.3.3 Boltzmann-Verteilung in Kraftfeldern
In konservativen Kraftfeldern verallgemeinert sich die Maxwell-Verteilung zur Boltzmann-Verteilung. Für eine potentielle Energie ist die Phasenraumverteilung:
wobei und die Dichte bei ist. Integration über die Geschwindigkeiten ergibt die räumliche Dichte:
Für die Gravitation wird dies zu:
Die Skalenhöhe charakterisiert den exponentiellen Abfall (z. B. ~8 km für die Erdatmosphäre). Die Maxwell-Boltzmann-Verteilung kombiniert beide Aspekte:
1.3.4 Äquipartitionstheorem und Energieverteilung
Das Äquipartitionstheorem besagt: Jeder quadratische Term in der Hamiltonfunktion eines Systems trägt zur mittleren Energie bei. Für eine Koordinate mit Energie ergibt die Boltzmann-Verteilung:
unter Verwendung von Gaußschen Integralen. Molekulare Freiheitsgrade umfassen:
Translation: 3 Terme →
Rotation: 2 (zweiatomig) oder 3 (mehratomig) Terme → oder
Vibration: Kinetische und potentielle Terme tragen jeweils pro Modus bei.
Für ein zweiatomiges Molekül beträgt die Gesamtenergie (starr) oder (nicht-starr). Die molare Wärmekapazität bei konstantem Volumen ist:
wobei die Anzahl der quadratischen Freiheitsgrade ist.
1.4 Mittlere freie Weglänge von Gasmolekülen
1.4.1 Mittlere Stoßfrequenz und mittlere freie Weglänge
Gasmoleküle bewegen sich mit hohen thermischen Geschwindigkeiten (z. B. Stickstoff bei 27 °C im Durchschnitt 476 m/s), dennoch erfolgt die makroskopische Diffusion langsam aufgrund häufiger Kollisionen, die molekulare Bahnen randomisieren. Die mittlere freie Weglänge quantifiziert die durchschnittliche Distanz, die ein Molekül zwischen Kollisionen zurücklegt, während die Stoßfrequenz die Anzahl der Kollisionen pro Zeiteinheit zählt. Zur Modellierung von Kollisionen werden Moleküle als starre Kugeln mit einem effektiven Durchmesser behandelt, wobei langreichweitige Anziehung ignoriert, aber kurzreichweitige Abstoßung berücksichtigt wird, die Überlappung verhindert.
Für identische Moleküle ist der Stoßquerschnitt . Wenn sich ein "Test"-Molekül mit einer durchschnittlichen Relativgeschwindigkeit durch ein Gas mit Teilchendichte bewegt, überstreicht es in der Zeit einen Stoßzylinder mit dem Volumen , wie in der folgenden Abbildung dargestellt:
Stoßzylinder eines Moleküls
Dann ist die Stoßfrequenz:
wobei die mittlere thermische Geschwindigkeit ist. Die mittlere freie Weglänge ergibt sich als:
Unter Verwendung des idealen Gasgesetzes wird dies zu:
Somit skaliert umgekehrt proportional zum Druck (z. B. Luft bei STP: m; bei Pa: m).
1.4.2 Verteilung der freien Weglängen
Die Wahrscheinlichkeit, dass ein Molekül eine Distanz ohne Kollision zurücklegt, folgt aufgrund zufälliger Kollisionen einem exponentiellen Abfall.
Angenommen, zum Zeitpunkt befinden sich Moleküle an Position , die noch keine Kollisionen hatten.
Nach einem Zeitintervall bewegt sich der Molekularstrahl zur Position , und Moleküle kollidieren und werden entfernt.
Das heißt, die Anzahl der Moleküle, deren freie Weglänge zwischen und liegt, ist .
Innerhalb des Distanzintervalls bis ist die Anzahl der Moleküle, die kollidieren, , proportional zur Anzahl der Moleküle bei , , und proportional zur Größe von . Die Proportionalitätskonstante ist , also:
Dies ergibt
wobei die Anzahl ist, die die Distanz überlebt. Die Wahrscheinlichkeitsdichte für eine freie Weglänge zwischen und ist:
Der Mittelwert stimmt mit überein. Diese Verteilung untermauert Phänomene wie die Abschwächung von Elektronenstrahlen in Vakuumröhren.
1.4.3 Viskosität
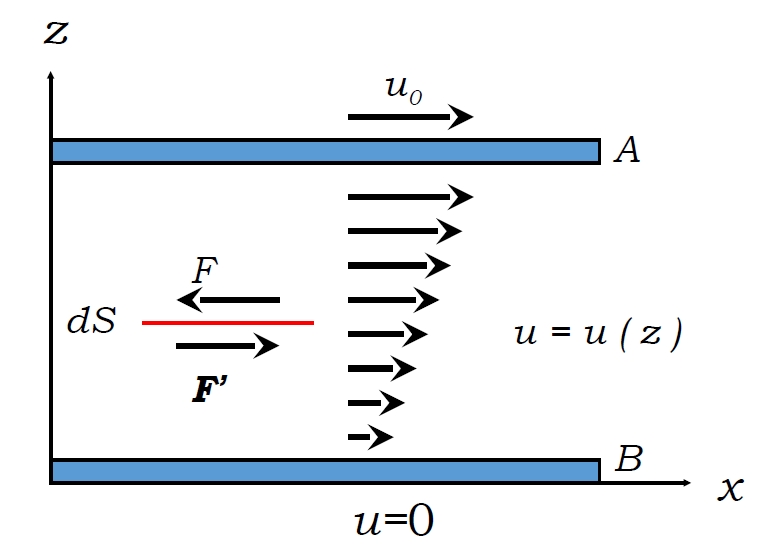
Wenn benachbarte Flüssigkeitsschichten sich mit unterschiedlichen Geschwindigkeiten bewegen (z. B. in einer -abhängigen Strömung), entsteht Viskosität durch Impulsübertragung senkrecht zur Strömungsrichtung.
Geschwindigkeitsgradient in geschichteter Strömung erzeugt viskose Kräfte
Newtonsches Viskositätsgesetz besagt:
wobei die dynamische Viskosität und der Geschwindigkeitsgradient ist. Die Kraft verlangsamt schnellere Schichten und beschleunigt langsamere, wodurch der Impuls ausgeglichen wird. Viskositätseinheiten sind Pa·s (Poise: 1 P = 0,1 Pa·s).
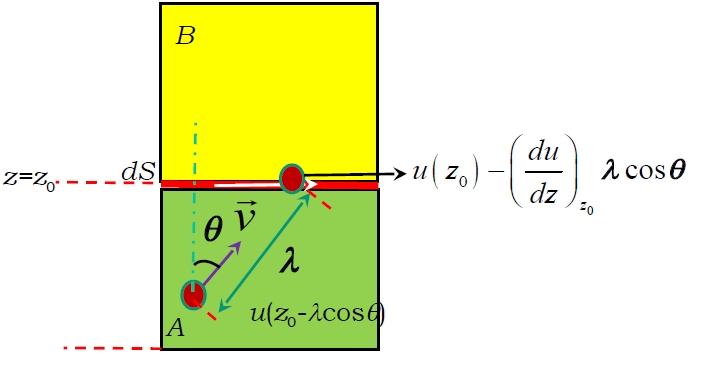
Hier ist eine Ableitung:
Überschüssiger Impuls
Die Anzahl der Moleküle mit Geschwindigkeit , die in der Zeit von Region A nach gelangen, ist
Der durch diese Moleküle erzeugte Impulsfluss ist
Der Impulsfluss, der von allen Molekülen von A nach B erzeugt wird, ist
Überschüssiger Impuls
Die Anzahl der Moleküle mit Geschwindigkeit , die in der Zeit von Region B nach gelangen, ist
Der durch diese Moleküle erzeugte Impulsfluss ist
Der Beitrag aller Moleküle von B nach A zum Impulsfluss ist
Der Beitrag zum Impulsfluss durch von allen Molekülen ist
Impuls im Fluid
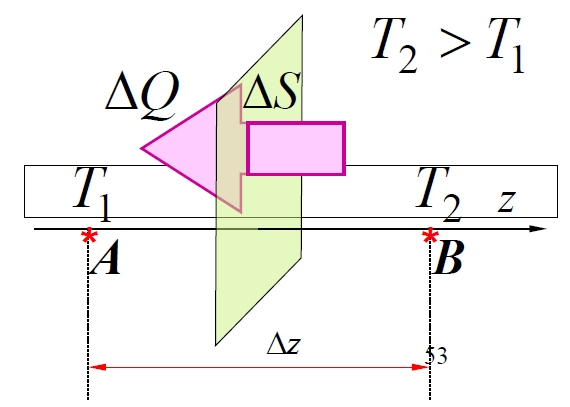
1.4.4 Wärmeleitung
Wärmeleitung tritt auf, wenn Temperaturgradienten vorhanden sind. Das Fouriersche Gesetz beschreibt den Wärmefluss durch die Fläche in der Zeit :
wobei die Wärmeleitfähigkeit (W·m⁻¹·K⁻¹) ist. Wärme fließt abwärts des Temperaturgradienten und überträgt Energie von heißen zu kalten Regionen. In Gasen dominiert die Leitung, wenn die Massenbewegung (Konvektion) vernachlässigbar ist.
Damit Wärmeleitung in Gasen ohne Druckunterschied () stattfinden kann, wird die Teilchendichte bestimmt durch:
Betrachten Sie die Regionen A (wärmer, bei ) und B (kälter, bei ) mit den Temperaturen und bzw.:
Unter Verwendung der mittleren Geschwindigkeit :
Für kleine Temperaturunterschiede ist der Fluss annähernd konstant:
Mikroskopisch erfolgt die Energieübertragung durch molekulare Kollisionen: Moleküle aus wärmeren Regionen tragen höhere kinetische Energie und tauschen diese bei Kollisionen mit Molekülen aus kälteren Regionen aus. Die durchschnittliche thermische Energie pro Molekül ist:
In Region A: (für translatorische Energie; für Moleküle mit Freiheitsgraden ist es , )
In Region B: ,
Die Anzahl der ausgetauschten Molekülpaare über eine Fläche in der Zeit wird geschätzt als:
(Der Faktor ergibt sich aus der Betrachtung des Bruchteils der Moleküle, die sich in einer bestimmten Richtung senkrecht zur Oberfläche in einem isotropen Gas bewegen).
Die entlang der positiven z-Achse pro ausgetauschtem Molekülpaar transportierte Nettenergie ist die Differenz:
Die gesamte Energie, die in der Zeit durch entlang der positiven z-Achse transportiert wird (d. h. die Wärme ), ist:
Der Temperaturunterschied steht im Zusammenhang mit dem Temperaturgradienten bei :
Dies einsetzend:
Vergleicht man dies mit dem Fourierschen Gesetz , identifiziert man die Wärmeleitfähigkeit als:
Unter Verwendung der molaren Wärmekapazität bei konstantem Volumen (wobei die Avogadro-Zahl ist) und der spezifischen Wärmekapazität (pro Masseneinheit) und der Dichte kann dies umgeschrieben werden als:
Demonstration der Wärmeleitung
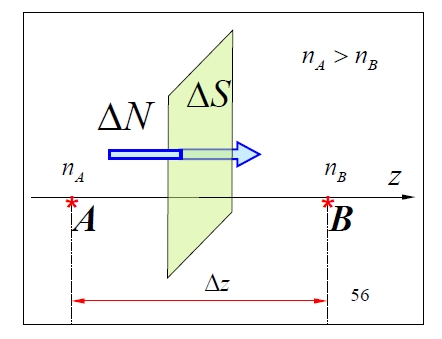
1.4.5 Diffusion
Diffusion transportiert Masse aufgrund von Dichtegradienten. Ficks Gesetz für die Masse , die in der Zeit die Fläche durchquert, lautet:
wobei der Diffusionskoeffizient (m²·s⁻¹) und die Massendichte ist. In Gasen entstehen Selbstdiffusion (identische Moleküle) und gegenseitige Diffusion (unterschiedliche Spezies) durch Nettomolekülfluss von Regionen hoher zu niedriger Dichte.
Betrachten Sie einen Druckunterschied, bei dem . Da der Druck über oder äquivalent bei konstanter Temperatur mit der Dichte zusammenhängt:
Die Nettomenge der Moleküle, die in der Zeit entlang der positiven -Achse durch transportiert wird, ist:
(Der Faktor berücksichtigt den Anteil der Moleküle, die senkrecht zur Oberfläche strömen).
Die entsprechende Nettomasse, die transportiert wird, ist:
Beziehung der Dichteunterschiede zum Gradienten bei :
Einsetzen:
Vergleich mit Ficks Gesetz ergibt den Diffusionskoeffizienten als:
Für Gemische unterschiedlicher Gase (gegenseitige Diffusion) hängt der Diffusionskoeffizient von den interagierenden Spezies ab. Ein Beispiel ist unten gezeigt:
Gas
(cm²·s⁻¹)
Gas 1: Wasserstoff ()
3
0.594
Gas 2: Kohlendioxid ()
1
0.605
Demonstration der Diffusion
1.5 Der Erste Hauptsatz der Thermodynamik
1.5.1 Thermodynamische Prozesse
Ein thermodynamischer Prozess oder einfach Prozess tritt auf, wenn sich der Zustand eines Systems im Laufe der Zeit ändert. Wenn beispielsweise ein vorgeschobener Kolben Gas in einem Zylinder komprimiert, ändern sich das Volumen, die Dichte, die Temperatur oder der Druck des Gases, und zu jedem Zeitpunkt während des Prozesses sind Dichte, Druck und Temperatur nicht im gesamten Gas identisch. Thermodynamische Prozesse werden in nicht-quasistatische Prozesse und quasistatische Prozesse eingeteilt.
Bei nicht-quasistatischen Prozessen geht das System von einem Gleichgewichtszustand in einen gestörten Zustand über, bevor es ein neues Gleichgewicht erreicht. Die Zeit von der Störung des Gleichgewichts bis zur Herstellung des neuen Gleichgewichts wird als Relaxationszeit () bezeichnet. Tatsächliche Prozesse verlaufen oft schnell, wobei das System weitere Änderungen durchläuft, bevor es ein neues Gleichgewicht erreicht, was bedeutet, dass das System Nichtgleichgewichtszustände durchläuft, die nicht durch Zustandsgrößen beschrieben werden können. Die freie Expansion eines idealen Gases ist ein typischer nicht-quasistatischer Prozess, bei dem "frei" bedeutet, dass das Gas ohne Widerstand expandiert.
Bei quasistatischen Prozessen ist jeder Zwischenzustand infinitesimal nah an einem Gleichgewichtszustand. Dies ist nur möglich, wenn der Prozess "unendlich langsam" verläuft. Für tatsächliche Prozesse erfordert die quasistatische Annäherung, dass die charakteristische Zeit der Zustandsänderung viel größer ist als die Relaxationszeit . Gleichgewichtszustände in quasistatischen Prozessen haben definierte Zustandsgrößenwerte, die für einfache Systeme durch Punkte in einem P-V-Diagramm dargestellt werden. Der quasistatische Änderungsprozess wird durch eine Kurve im P-V-Diagramm dargestellt, die als Prozesskurve bezeichnet wird. Obwohl eine ideale Grenze, sind quasistatische Prozesse für die Thermodynamik grundlegend und praktisch bedeutsam. Sofern nicht anders angegeben, beziehen sich thermodynamische Prozesse auf quasistatische Prozesse. Zum Beispiel ist das allmähliche Erhitzen eines Systems von auf durch kleine Temperaturerhöhungen quasistatisch, während der direkte Kontakt eines Systems bei mit einer Wärmequelle bei dies nicht ist.
1.5.2 Arbeit
Arbeit ist eine Methode des Energieaustauschs.
In der Thermodynamik repräsentiert sie die Umwandlung zwischen geordneter mechanischer Energie aus der Umgebung und ungeordneter thermischer Energie des Systems, bezeichnet als .
Arbeit kann den Zustand des Systems ändern und nicht nur mechanische Bewegung, sondern auch thermische Bewegung und elektromagnetische Zustände beeinflussen, wie z. B. Reibungserwärmung (mechanische Arbeit) oder elektrische Erwärmung (elektrische Arbeit).
Für Gase, die quasistatische Prozesse durchlaufen, ist die vom System verrichtete Arbeit gegeben durch
und
wobei Arbeit am System anzeigt. Grafisch entspricht die Arbeitsgröße der Fläche unter der Prozesskurve in einem -Diagramm. Arbeit ist keine Zustandsgröße, sondern eine Prozessgröße – sie hängt vom Weg zwischen den Zuständen ab. Für verschiedene Prozesse vom Gleichgewichtszustand 1 zu 2 variiert die von der Umgebung verrichtete Arbeit. Arbeit ist eine Prozessgröße, wegeabhängig.
1.5.3 Wärme
Wärme ist eine weitere Methode zur Zustandsänderung eines Systems, die sich von Arbeit unterscheidet. Während Arbeit Energieübertragung durch generalisierte Kräfte verursacht, die generalisierte Verschiebungen hervorrufen, erfolgt Wärmeübertragung aufgrund von Temperaturunterschieden. Joules Experimente zeigten, dass Wärmeproduktion oder -verschwinden immer mit äquivalentem Verschwinden oder Produzieren anderer Energieformen (mechanisch, elektrisch) einhergeht, was beweist, dass keine separat konservierte "Caloric"-Substanz existiert. Stattdessen konservieren Wärme, mechanische Energie und elektrische Energie zusammen die Energie. Wärme () ist übertragene Energie: positiv, wenn sie vom System absorbiert wird, negativ, wenn sie abgegeben wird. Sowohl Wärme als auch Arbeit quantifizieren Energieänderungen, sind prozessabhängig, unterscheiden sich aber in ihrem Ursprung: Arbeit stammt aus mechanischen Wechselwirkungen, Wärme aus thermischen Wechselwirkungen.
1.5.4 Der Erste Hauptsatz der Thermodynamik
Joule's Experimente enthüllten eine eindeutige Äquivalenz zwischen Wärme und Arbeit (1 cal = 4,186 J) und zeigten die Umwandlung zwischen mechanischer/elektromagnetischer und thermischer Bewegung. Dies führte zum Gesetz der Energieerhaltung und -umwandlung: Alle Materie besitzt Energie in verschiedenen Formen, die untereinander umwandelbar und zwischen Objekten übertragbar sind, wobei die Gesamtmenge erhalten bleibt. Eine alternative Aussage: Perpetuum mobile erster Art sind unmöglich.
Für ein System, das von einem Anfangszustand 1 zu einem Endzustand 2 über einen adiabatischen Prozess (kein Wärmeaustausch) wechselt, ist die von der Umgebung verrichtete Arbeit die adiabatische Arbeit. Joule's Experimente zeigten, dass für feste Anfangs- (Zustand 1, Temperatur ) und Endzustände (Zustand 2, Temperatur ) die adiabatische Arbeit wegunabhängig ist. Somit wird die innere Energie als Zustandsfunktion definiert, die für jeden adiabatischen Prozess zwischen den Zuständen 1 und 2 erfüllt.
Für nicht-adiabatische Prozesse mit Arbeit , die am System verrichtet wird, und absorbierter Wärme ergibt sich aus der Energieerhaltung:
Dies ist der mathematische Ausdruck des ersten Hauptsatzes, wobei für Arbeit am System und für absorbierte Wärme gilt. Für infinitesimale Prozesse:
Hier ist exakt (Zustandsfunktion), während und inexakte Differentiale (Prozessgrößen) sind. Der erste Hauptsatz gilt sowohl für quasistatische als auch für nicht-quasistatische Prozesse, obwohl Anfangs-/Endzustände für Berechnungen im Gleichgewicht sein müssen. Wenn nur Volumenarbeit existiert:
1.5.5 Wärmekapazität und Enthalpie
Die Wärmekapazität ist definiert als
wenn sich die Temperatur eines Systems um erhöht, während ihm die Wärmemenge zugeführt wird. Sie hängt vom Prozess, der Substanz und der Masse ab.
Bei konstantem Volumen () ist , also , was zur Wärmekapazität bei konstantem Volumen führt:
wobei und Funktionen von und sind.
Bei konstantem Druck ist die Arbeit , also ist die absorbierte Wärme:
Definiert man die Enthalpie (Zustandsfunktion), so ist . Für infinitesimale Prozesse:
Somit ist die Wärmekapazität bei konstantem Druck:
wobei und Funktionen von und sind. Für ideale Gase gilt .
1.5.6 Innere Energie von Gasen und Joule-Thomson-Experiment
In Joules freiem Expansionsversuch von 1845 dehnte sich Gas in ein Vakuum aus, ohne dass eine Temperaturänderung beobachtet wurde. Die Anwendung des ersten Hauptsatzes (, ) ergibt , was auf eine konstante innere Energie während der adiabatischen freien Expansion hindeutet. Daher gilt für ideale Gase:
Joules Experiment war jedoch ungenau, da die große Wärmekapazität von Wasser die Temperaturänderungen des Gases maskierte. Das Joule-Thomson-Experiment (1852) verbesserte die Genauigkeit durch Untersuchung der adiabatischen Drosselung.
Joule-Thomson-Experiment
Bei der adiabatischen Drosselung strömt Gas mit hohem Druck durch einen porösen Stopfen zu niedrigem Druck, ohne Wärmeaustausch ().
Die Prozessgleichung ergibt eine konstante Enthalpie. Der Joule-Thomson-Koeffizient:
bestimmt die Temperaturänderung: für Kühlung, für Erwärmung. Reale Gase haben , was beweist, dass die innere Energie sowohl von der Temperatur als auch vom Volumen abhängt und molekulare Kräfte vorhanden sind. Dieser Effekt ermöglicht die praktische Verflüssigung von Gasen (z. B. Linde-Verfahren).
Für ideale Gase, die und erfüllen:
Die innere Energie hängt ausschließlich von der Temperatur ab, da zwischenmolekulare Kräfte vernachlässigbar sind. Nach dem Äquipartitionstheorem trägt jeder Freiheitsgrad pro Mol bei. Für Freiheitsgrade:
Die Wärmekapazität bei konstantem Volumen ergibt sich durch Differentiation:
Die Enthalpie ist definiert als . Einsetzen des idealen Gasgesetzes :
Differentiation nach bei konstantem Druck:
Somit ist die molare Wärmekapazität bei konstantem Druck:
Das Verhältnis der Wärmekapazitäten ist:
Dies bestätigt und für alle .
Prozessanwendungen:
Isochor ( konstant): , , , .
Isobar ( konstant):
Isotherm ( konstant): , , .
Adiabatisch (): , was zu , , führt. Arbeit .
Für einen polytropen Prozess wird die molare Wärmekapazität aus dem ersten Hauptsatz und der Prozessgleichung abgeleitet. Für 1 Mol ideales Gas:
Die molare Wärmekapazität ist definiert als , also:
Aus der polytropen Gleichung und dem idealen Gasgesetz lösen wir nach auf:
Differenzieren nach :
Umstellen, um auszudrücken:
Einsetzen:
Somit:
Unter Verwendung von und drücken wir aus als:
Einsetzen in (2):
Lösen nach in Bezug auf Wärmekapazitäten:
Dann können Spezialfälle überprüft werden:
Isobar ():
Isochor (): (Grenzwert von )
Isotherm (): (undefiniert, konsistent mit )
Adiabatisch (): (da )
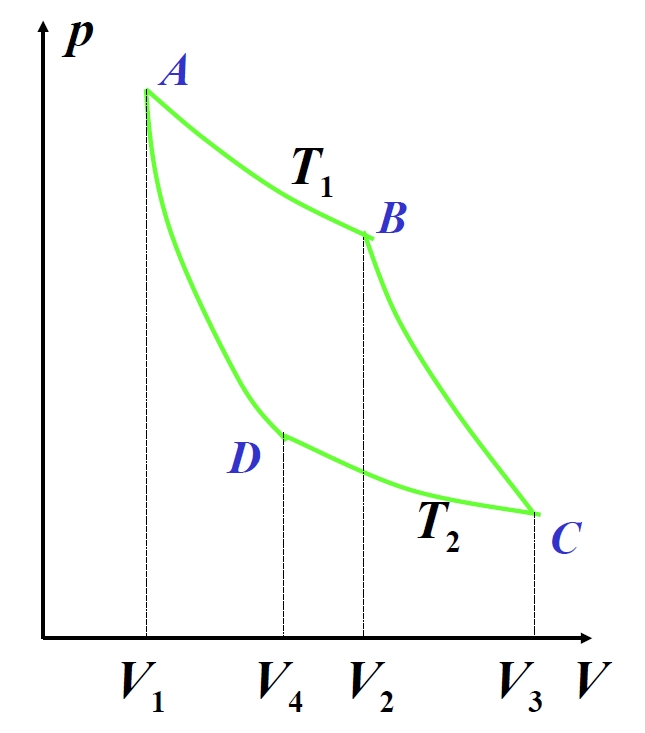
1.5.7 Zyklische Prozesse und Carnot-Zyklus
Zyklische Prozesse beinhalten eine Arbeitsmasse, die nach Abschluss einer Reihe von thermodynamischen Änderungen zu ihrem Anfangszustand zurückkehrt. Quasistatische Zyklen werden als geschlossene Kurven in P-V-Diagrammen dargestellt, wobei Uhrzeigersysteme als Wärmekraftmaschinen und Gegenuhrzeigersysteme als Kühlsysteme fungieren. Bei Wärmekraftmaschinen absorbiert die Arbeitsmasse Wärme von einem Hochtemperaturreservoir bei , verrichtet Nettoarbeit an der Umgebung und gibt Wärme an ein Niedertemperaturreservoir bei ab. Der erste Hauptsatz ergibt:
mit der thermischen Effizienz definiert als:
P-V-Diagramm des Carnot-Diagramms
Der Carnot-Zyklus – ein fundamentales Modell für Wärmekraftmaschinen – kombiniert zwei isotherme und zwei adiabatische Prozesse. Für eine ideale Gas-Arbeitsmasse:
Isotherme Expansion (A→B):
Adiabatische Expansion (B→C):
Isotherme Kompression (C→D):
Adiabatische Kompression (D→A):
Die adiabatischen Gleichungen ergeben die Volumenverhältnisbeziehung:
Einsetzen in die Effizienzformel:
Dieses Ergebnis hängt ausschließlich von den Reservoirtemperaturen ab und ist unabhängig von der Arbeitsmasse.
Für Kühlsystem-Zyklen (gegen den Uhrzeigersinn im P-V-Diagramm) absorbiert die Arbeitsmasse Wärme von einem Kaltreservoir () unter Arbeitsaufwand und gibt Wärme an ein Warmreservoir () ab. Der Leistungskoeffizient ist:
Für Carnot-Kühlsysteme ist die maximal mögliche Leistung:
Otto-Zyklus (konstante Volumen-Erwärmung) mit der Effizienz:
wobei das Verdichtungsverhältnis ist.
Diesel-Zyklus (konstante Druck-Erwärmung) mit höherer Effizienz aufgrund größerer Verdichtungsverhältnisse.
1.6 Der Zweite Hauptsatz der Thermodynamik
1.6.1 Der Zweite Hauptsatz der Thermodynamik
Der Zweite Hauptsatz der Thermodynamik befasst sich mit der Richtung natürlicher Prozesse und ergänzt das Energieerhaltungsprinzip des Ersten Hauptsatzes. Während der Erste Hauptsatz Perpetuum mobile erster Art (Verletzung der Energieerhaltung) verbietet, schränkt er die Prozessrichtung nicht ein. Zum Beispiel fließt Wärme spontan von hoher zu niedriger Temperatur, aber nicht umgekehrt. Der Zweite Hauptsatz löst dies durch zwei äquivalente Formulierungen:
Kelvin-Aussage (1851): Es ist unmöglich, Wärme vollständig aus einer einzigen Wärmequelle in Arbeit umzuwandeln, ohne andere Effekte. Dies impliziert, dass Wärmekraftmaschinen keine 100%ige Effizienz erreichen können () und verbietet Perpetuum mobile zweiter Art.
Clausius-Aussage (1850): Wärme kann nicht spontan von einem kalten zu einem heißen Körper fließen, ohne externe Arbeitszufuhr. Dies etabliert die gerichtete Natur des Wärmetransfers und begrenzt die Effizienz von Kühlsystemen ().
Thermodynamische Prozesse weisen aufgrund inhärenter Ungleichgewichte Irreversibilität auf. Ein Prozess ist nur dann reversibel, wenn sowohl das System als auch die Umgebung zu ihren Anfangszuständen ohne Nettoänderungen zurückkehren. Kelvins Aussage offenbart die Irreversibilität der Arbeits-zu-Wärme-Umwandlung, während Clausius' Aussage die Irreversibilität bei der Wärmeübertragung zeigt. Diese Aussagen sind logisch äquivalent: Die Verletzung einer impliziert die Verletzung der anderen. Entscheidend ist, dass alle irreversiblen Prozesse miteinander verbunden sind – was zeigt, dass Irreversibilität in einem Prozess (z. B. freie Expansion) Irreversibilität in anderen (z. B. Wärmeübertragung) impliziert. Das Kernprinzip ist, dass alle makroskopischen thermischen Prozesse irreversibel sind; reversible Prozesse sind Idealisierungen, die quasistatische, dissipationsfreie Bedingungen erfordern.
Nun beweisen wir die Äquivalenz zwischen Kelvin- und Clausius-Aussagen:
(1) Clausius falsch ⇒ Kelvin falsch
Angenommen, Gerät verletzt Clausius: überträgt Wärme von einem Kaltreservoir zu einem Warmreservoir ohne Arbeitszufuhr. Kombinieren Sie dies mit einer reversiblen Wärmekraftmaschine , die zwischen und arbeitet:
Nettoeffekt: Vollständige Umwandlung von Wärme von in Arbeit , was Kelvin verletzt.
(2) Kelvin falsch ⇒ Clausius falsch
Angenommen, Gerät verletzt Kelvin: wandelt Wärme von vollständig in Arbeit um. Betreiben Sie ein Kühlsystem mit dieser Arbeit:
Nettoeffekt: Wärme fließt spontan von nach , was Clausius verletzt.
Irreversibilität entsteht statistisch aus molekularer Unordnung. Betrachten Sie Gasmoleküle in einem geteilten Behälter: Nach Entfernung der Trennwand verteilen sich die Moleküle gleichmäßig. Die gleichmäßige Verteilung hat die höchste thermodynamische Wahrscheinlichkeit (), während geordnete Zustände (z. B. alle Moleküle auf einer Seite) für große vernachlässigbare haben. Das Boltzmann-Postulat besagt, dass isolierte Systeme im Gleichgewicht gleiche mikroskopische Zustands wahrscheinlichkeiten haben. Natürliche Prozesse entwickeln sich hin zu makroskopischen Zuständen mit höherem , was die Unordnung erhöht. Die statistische Interpretation des Zweiten Hauptsatzes: Isolierte Systeme entwickeln sich von Zuständen niedriger zu Zuständen hoher , was eine erhöhte Zufälligkeit widerspiegelt. Dies gilt nur für makroskopische Systeme, nicht für mikroskopische Phänomene wie die Brownsche Bewegung, und ist auf endliche isolierte Systeme beschränkt, nicht auf das Universum.
1.6.2 Carnot-Theorem
Das Carnot-Theorem legt die theoretischen Grenzen für die Effizienz von Wärmekraftmaschinen fest, die zwischen thermischen Reservoirs bei Temperaturen (hoch) und (niedrig) arbeiten. Das Theorem besagt: Alle reversiblen Maschinen, die zwischen diesen Reservoirs arbeiten, erreichen die identische Effizienz
während irreversible Maschinen
erfüllen. Der Beweis für die Gleichheit reversibler Maschinen erfolgt durch Widerspruch. Betrachten Sie zwei reversible Maschinen und , die zwischen Reservoirs bei und arbeiten. Angenommen, , dann setzen wir reversibel als Kühlsystem mit angepassten Zyklen ein, die und erfüllen. Die Ungleichung impliziert
wobei und . Das zusammengesetzte System entzieht Nettoarbeit und tauscht keinen Netto-Wärmeaustausch mit beiden Reservoirs aus. Dies verletzt die Kelvin-Aussage, indem Wärme vollständig in Arbeit umgewandelt wird, ohne andere Effekte, was erzwingt
Für irreversible Maschinen sei irreversibel und reversibel zwischen und . Angenommen, und betreiben Sie als Kühlsystem mit angepasstem , impliziert die Ungleichung
wobei und . Das zusammengesetzte System entzieht Nettoarbeit bei null Netto-Wärmeaustausch bei und Netto-Wärmeverlust bei . Wenn , verletzt dies die Kelvin-Aussage, indem positive Arbeit aus nicht-positiver Wärmeentnahme erzeugt wird, was beweist
mit Gleichheit nur für reversible Maschinen.
1.6.3 Thermodynamische Temperaturskala
Die thermodynamische Temperaturskala, etabliert von Lord Kelvin unter Verwendung des Carnot-Theorems, liefert eine universelle Temperaturdefinition, die unabhängig von Materialeigenschaften ist. Für eine reversible Wärmekraftmaschine, die zwischen Reservoirs mit empirischen Temperaturen und arbeitet, erfüllt das Wärme-Verhältnis
wobei eine universelle Funktion ist. Einführung eines dritten Reservoirs bei mit einer Hilfs-Reversibelmaschine ergibt
Die Kombination dieser Verhältnisse ergibt
wobei die universelle Funktion nur von der Temperatur abhängt. Die Definition der thermodynamischen Temperatur etabliert
Die Festlegung des Tripelpunkts von Wasser auf stimmt diese Skala mit dem idealen Gasthermometer überein, wo anwendbar, und bestätigt
Diese Ableitung liefert die theoretische Grundlage für die absolute Temperaturskala, die in der Carnot-Effizienzformel verwendet wird
1.6.4 Entropie
Der Zweite Hauptsatz der Thermodynamik befasst sich mit der Richtung natürlicher Prozesse und ergänzt das Energieerhaltungsprinzip des Ersten Hauptsatzes. Kelvins Aussage (1851) erklärt, dass es unmöglich ist, Wärme vollständig aus einer einzigen Quelle in Arbeit umzuwandeln, ohne andere Effekte, was impliziert, dass Wärmekraftmaschinen keine 100%ige Effizienz erreichen können () und Perpetuum mobile zweiter Art verbietet. Clausius' Aussage (1850) stellt fest, dass Wärme nicht spontan von kalten zu heißen Objekten fließen kann, ohne Arbeitsaufwand, was die gerichtete Natur des Wärmetransfers definiert und die Effizienz von Kühlsystemen begrenzt (). Thermodynamische Prozesse weisen inhärente Irreversibilität aufgrund von Ungleichgewichten auf, wobei reversible Prozesse erfordern, dass sowohl das System als auch die Umgebung exakt zu ihren Anfangszuständen zurückkehren – eine Idealisierung, die quasistatische, dissipationsfreie Bedingungen erfordert. Kelvins Aussage offenbart die Irreversibilität der Arbeits-zu-Wärme-Umwandlung, während Clausius' Aussage die Irreversibilität der Wärmeübertragung demonstriert, wobei beide Formulierungen logisch äquivalent sind: Die Verletzung einer impliziert die Verletzung der anderen. Wenn beispielsweise die Wärmeübertragung reversibel wäre, könnte man ein System konstruieren, das Wärme von kalten zu heißen Reservoirs ohne Arbeit überträgt, was gegen Clausius' Aussage verstößt. Ebenso führt die Annahme der Reversibilität der freien Expansion zur Verletzung von Kelvins Aussage und bestätigt, dass alle makroskopischen thermischen Prozesse grundsätzlich irreversibel sind.
Irreversibilität entsteht statistisch aus molekularer Unordnung. Betrachten Sie Gasmoleküle in einem geteilten Behälter: Nach Entfernung der Trennwand verteilen sich die Moleküle gleichmäßig mit maximaler thermodynamischer Wahrscheinlichkeit , während geordnete Zustände (z. B. alle Moleküle auf einer Seite) für vernachlässigbar werden. Boltzmanns Postulat besagt, dass isolierte Gleichgewichtssysteme gleiche mikroskopische Zustands wahrscheinlichkeiten haben, was erklärt, warum natürliche Prozesse sich zu Zuständen höherer entwickeln und die Unordnung erhöhen. Diese statistische Interpretation – isolierte Systeme entwickeln sich von Zuständen niedriger zu Zuständen hoher – gilt ausschließlich für makroskopische Systeme und endliche isolierte Umgebungen. Das Carnot-Theorem legt Effizienzgrenzen zwischen Reservoirs bei fest: Alle reversiblen Maschinen erreichen , während irreversible Maschinen erfüllen. Der Beweis für die Gleichheit reversibler Maschinen beinhaltet den Nachweis, dass zwei Maschinen mit unterschiedlichen Effizienzen bei Kopplung Nettowärme aus einem einzelnen Reservoir entziehen würden, was gegen Kelvins Aussage verstößt.
Kelvin etablierte eine universelle Temperaturskala unter Verwendung des Carnot-Theorems. Für eine reversible Wärmekraftmaschine zwischen Reservoirs:
nachdem die thermodynamische Temperatur durch Wärme-Verhältnisse definiert und der Tripelpunkt von Wasser bei festgelegt wurde. Die Entropie wird über reversible Zyklen definiert:
Kombination mit dem Ersten Hauptsatz ergibt:
Für ideale Gase:
Die Clausius-Ungleichung impliziert , was zum Prinzip der Entropieerhöhung für isolierte Systeme führt:
wobei die freie Expansion und die Wärmeübertragung ergibt. Boltzmanns mikroskopische Interpretation:
quantifiziert die Unordnung durch die Anzahl der Mikrozustände . Für offene Systeme:
wobei das Maxwellsche Dämonen-Paradoxon durch Landauers Prinzip gelöst wird: das Löschen von 1 Bit dissipiert mindestens Wärme.
Teil II. Klassische Statistische Mechanik
Hinweis:
Bevor Sie diesen Teil lesen, stellen Sie bitte sicher, dass Sie Klassische Mechanik, Quantenmechanik und Wahrscheinlichkeit gelernt haben.
In Teil II und Teil III werden die Boltzmann-Konstanten alle auf normiert. Daher haben Temperatur und Energie die gleiche Einheit.
2.1 Beschreibung von Zuständen
Die statistische Physik verbindet das Verhalten einzelner Teilchen mit den messbaren Eigenschaften von Materialien, die wir im Alltag beobachten. Um zu verstehen, wie diese Verbindung funktioniert, benötigen wir zunächst eine Möglichkeit, den detaillierten Zustand eines Vielteilchensystems zu beschreiben.
Betrachten Sie ein isoliertes System, das aus Teilchen besteht, wie Moleküle in einem verschlossenen Behälter. Jedes Teilchen benötigt sechs Zahlen, um seinen mechanischen Zustand vollständig zu spezifizieren: drei Ortskoordinaten und drei Impulskomponenten . Für das gesamte System müssen wir alle Teilchen gleichzeitig berücksichtigen.
Der vollständige mikroskopische Zustand – genannt Mikrozustand – wird daher durch Auflistung aller Positionen und Impulse zusammen beschrieben:
Diese Sammlung von Zahlen bildet einen mathematischen Punkt in einem -dimensionalen abstrakten Raum, der als Phasenraum bekannt ist.
Die klassische Mechanik zeigt, dass wir, wenn wir diesen Phasenraum-Punkt zu einem beliebigen Zeitpunkt kennen und die Hamilton-Funktion des Systems (allgemein als Energie betrachtet), prinzipiell seine zukünftige Entwicklung durch die Hamiltonschen Gleichungen bestimmen können:
.
Somit definiert ein einzelner Punkt im Phasenraum vollständig den momentanen mechanischen Zustand des gesamten Systems.
Für makroskopische Systeme, bei denen ist, wird die Verfolgung einzelner Phasenraum-Punkte jedoch prinzipiell unmöglich. Darüber hinaus mitteln Labor-Messungen von Größen wie Druck oder Temperatur inhärent über eine enorme Anzahl von Mikrozuständen – ungefähr Mikrozustände tragen zu einer einzigen Sekunde makroskopischer Beobachtung bei.
Diese praktische Einschränkung erzwingt eine Verlagerung von der deterministischen Mechanik zur probabilistischen Beschreibung. Anstatt exakten Trajektorien zu folgen, müssen wir betrachten, wie Mikrozustände im Phasenraum verteilt sind, was uns zur Kernmethodik der statistischen Physik führt.
Basierend auf dieser Beschreibung müssen wir die Charakterisierung des Makrosystems spezifizieren. Für ein isoliertes System (kein Austausch von Teilchen und Energie mit der Umgebung) kann sein Makrozustand vollständig durch 3 messbare Erhaltungsgrößen bestimmt werden:
Teilchenzahl
Volumen des Systems
Energie des Systems
Warum diese drei? Makroskopische Messungen können Mikrozustände, die dasselbe teilen, nicht unterscheiden. Daher bestimmen die Parameter einen Makrozustand eines Systems. Ein solcher Makrozustand enthält eine massive Anzahl von Mikrozuständen. Er entspricht einer Menge im Phasenraum:
Die Bedingung definiert eine -dimensionale Hyperebene (Volumen Null), was Schwierigkeiten bei der Berechnung der Wahrscheinlichkeit verursacht. Um dieses Problem zu lösen, müssen wir die Energieschale einführen.
Basierend auf der Hyperebene erweitern wir eine weitere Dimension, um ihr eine kleine “Dicke” zu geben. Dann wird die Hyperebene zu einem dünnen Raum, dargestellt durch:
Dieser erweiterte Raum wird als Energieschale bezeichnet. Beachten Sie, dass kleiner als sein muss, aber es ist notwendig, ausreichend Mikrozustände einzuschließen ().
Mit einer Dimension von können wir die Wahrscheinlichkeit im Phasenraum berechnen.
Wir können das “Volumen” der Energieschale ermitteln:
wobei
Das Volumen beschreibt die Anzahl der Mikrozustände in .
2.2 Boltzmann-Postulat
Anfänglich versuchten Forscher, Verbindungen zwischen Makro- und Mikrozuständen durch die ergodische Annahme herzustellen. Sie glaubten, dass isolierte Systeme alle erreichbaren Zustände auf einer Energiehyperebene über eine ausreichend lange Zeit durchlaufen. Die Zeitspanne, die für eine strikte Durchquerung erforderlich ist, ist jedoch weit größer als das Alter des Universums, sodass sie messtechnisch unmöglich ist.
Um dieses Problem zu lösen, stellte Boltzmann das wichtigste Postulat der statistischen Mechanik auf:
Boltzmann-Postulat: Für ein isoliertes System im Gleichgewicht haben alle Mikrozustände gleiche Wahrscheinlichkeit.
Gleichgewicht bedeutet, dass alle beobachtbaren Eigenschaften (z. B. Temperatur, Druck, Dichte) zeitinvariant und räumlich homogen sind. Basierend auf diesem Postulat können wir die Wahrscheinlichkeit eines Mikrozustands in einer Energieschale ermitteln:
wobei der Mikrozustand ist.
Fast alles in der statistischen Mechanik basiert auf Boltzmanns Postulat.
2.3 Entropie
2.3.1 Gibbs-Shannon-Entropie
Da makroskopische Messungen Mikrozustände nicht unterscheiden können, während ein Makrozustand massive Mikrozustände einschließt, benötigen wir eine Größe, um das Ausmaß der Ununterscheidbarkeit zu quantifizieren.
Stellen Sie sich vor, Sie sagen das Wetter für morgen voraus: Wenn bekannt ist, dass morgen definitiv sonnig sein wird (), gibt es keinen Zweifel am Ergebnis der Vorhersage und die Unsicherheit ist Null. Wenn Sie erfahren, dass es sonnig sein wird, ist die erhaltene Informationsmenge ebenfalls fast Null (Sie wussten es bereits). Wenn die Wahrscheinlichkeit, dass es morgen sonnig oder regnerisch ist, 50/50 beträgt (), ist die Vorhersage am unsichersten. Sie erhalten die meiste Information (eliminieren die meiste Unsicherheit), wenn Sie erfahren, dass es sonnig (oder regnerisch) sein wird. Wenn die Wahrscheinlichkeit, dass es morgen sonnig ist, 0,9 und regnerisch 0,1 beträgt, liegt die Unsicherheit irgendwo dazwischen. Die Information, die das sonnige Wetter bringt, ist (erwartungsgemäß) geringer, und die Information, die das regnerische Wetter bringt, ist größer (unerwartet).
Aus dem obigen Beispiel ist klar, dass die von den Ereignissen getragene Information stark mit der Wahrscheinlichkeitsverteilung zusammenhängt. Im Allgemeinen können wir eine Funktion definieren, um die Informationsmenge zu beschreiben. Diese Funktion sollte die folgenden Bedingungen erfüllen:
ist stetig
Wenn ein Ereignis mit Wahrscheinlichkeit eintritt,
Für unabhängige Ereignisse , ,
Wir leiten nun die explizite Form von ab:
In Bezug auf Wahrscheinlichkeiten, wobei und , haben wir die funktionale Gleichung:
Definieren Sie für . Aus den Bedingungen ist stetig, , und erfüllt:
Wir nehmen an, dass auf differenzierbar ist. Diese Annahme ist gerechtfertigt, da die Stetigkeit von und die funktionale Gleichung implizieren, dass differenzierbar ist (wie üblich bei der Lösung von Cauchy’schen Funktionalgleichungen).
Fixieren Sie ein beliebiges . Betrachten Sie die funktionale Gleichung als Funktion von :
Differenzieren Sie beide Seiten nach (wobei als konstant betrachtet wird):
Linke Seite: nach der Kettenregel.
Rechte Seite: , da konstant in Bezug auf ist.
Somit:
Lösen Sie nach :
Setzen Sie die Ausdrücke für gleich:
Umstellen:
Gleichung (*) gilt für alle . Da die linke Seite nur von und die rechte Seite nur von abhängt, müssen beide Seiten einer Konstanten, sagen , gleich sein. Daher:
wobei eine Konstante ist. Lösen Sie nach :
Integrieren Sie beide Seiten nach :
wobei die Integrationskonstante ist.
Wenden Sie die Bedingung an:
Da , haben wir . Somit:
Wenden Sie nun die Bedingung für alle an. Für ist , also um sicherzustellen, muss gelten. Sei , wobei . Dann:
Dies ist äquivalent zu:
da jede Logarithmusbasis in die Konstante absorbiert werden kann (da , also entsprechend skaliert).
Nun haben wir bewiesen, dass . Zurück zum Kontext der Mikrozustände können wir die Gibbs-Shannon-Entropie definieren:
wobei die Wahrscheinlichkeitsdichte im Phasenraum ist.
Die Gibbs-Shannon-Entropie spiegelt die Unsicherheit und Unordnung eines thermodynamischen Systems wider.
2.3.2 Boltzmann-Entropie
Nach der Gibbs-Shannon-Entropie können wir eine weitere Art von Entropie definieren, die Boltzmann-Entropie genannt wird.
wobei das Volumen des Phasenraums ist. Wir können feststellen, dass wenn , . Die Boltzmann-Entropie ist also ein Spezialfall der Gibbs-Shannon-Entropie im Gleichgewicht. Übrigens ist die Boltzmann-Entropie nur im Gleichgewicht definiert, während die Gibbs-Shannon-Entropie auch außerhalb des Gleichgewichts definiert werden kann.
2.3.3 Prinzip der maximalen Entropie
Boltzmanns Postulat der gleichen Wahrscheinlichkeit führt zum Prinzip der maximalen Entropie: Die Gibbs-Shannon-Entropie eines isolierten thermodynamischen Systems ist im Gleichgewicht maximiert. Der Prozess der Konvergenz zum Gleichgewicht ist ein Prozess der Entropiezunahme.
Es gibt einen Beweis:
Das Gleichheitszeichen gilt, wenn . Dieses Prinzip bedeutet, dass Gleichgewichtszustände maximale Entropie ergeben. Gemäß dem 2. Hauptsatz der Thermodynamik tendieren alle isolierten Systeme dazu, sich in Richtung des Gleichgewichtszustands zu entwickeln.
2.4 Ensembles
Ein Ensemble in der statistischen Mechanik ist eine Wahrscheinlichkeitsverteilung über den Phasenraum des Systems, die unser Wissen (oder Unwissen) über den exakten Mikrozustand des Systems repräsentiert.
2.4.1 Mikrokanonisches Ensemble
Das mikrokanonische Ensemble ist der Ausgangspunkt der statistischen Gleichgewichtsmechanik. Es beschreibt ein isoliertes System – eines, das weder Energie, Volumen noch Teilchen mit seiner Umgebung austauschen kann – und weist seinen mikroskopischen Zuständen Wahrscheinlichkeiten zu.
In der klassischen Mechanik wird der Zustand eines Systems von Teilchen, das in einer Box mit dem Volumen eingeschlossen ist, durch einen Punkt im -dimensionalen Phasenraum der Positionen und Impulse spezifiziert. Die Thermodynamik bezieht sich jedoch nicht auf Mikrozustände – sie befasst sich nur mit makroskopischen Variablen wie Energie , Volumen und Teilchenzahl . Das Ziel der statistischen Mechanik ist es, die Thermodynamik als Konsequenz statistischer Eigenschaften mikroskopischer Freiheitsgrade zu erklären.
Erinnern Sie sich an die Energieschale . Anstelle der charakteristischen Funktion können wir sie auch mit der Delta-Funktion darstellen:
wobei als “Oberflächeninhalt” der konstanten Energiefläche bezeichnet wird.
Im mikrokanonischen Ensemble ist die fundamentale thermodynamische Größe die Boltzmann-Entropie (im Gleichgewicht), die definiert ist als:
Der Faktor im Nenner dient zur Eliminierung quantenmechanischer Effekte. Wir können die Entropie umschreiben:
Es ist offensichtlich, dass und beide extensiv sind, während subextensiv ist. Daher können wir im thermodynamischen Limes ganz ignorieren und haben:
In der Thermodynamik können alle Zustandsvariablen aus der fundamentalen Relation abgeleitet werden. Es ist nützlich, sich diese Funktion als eine Oberfläche in einem vierdimensionalen Raum vorzustellen, der von aufgespannt wird. Diese Oberfläche wurde von Callen als fundamentale Oberfläche bezeichnet. Das mikrokanonische Ensemble realisiert diese Idee konkret, indem es die Entropie mit dem Volumen des Phasenraums verbindet. Sobald wir die Entropie als Funktion von Energie, Volumen und Teilchenzahl haben, können wir alle anderen thermodynamischen Größen als ihre partiellen Ableitungen definieren.
Temperatur ist definiert als:
Die Temperatur misst, wie schnell die Anzahl der zugänglichen Mikrozustände mit der Energie zunimmt. Damit die Temperatur wohldefiniert und positiv ist, muss die Funktion monoton mit wachsen. Physikalisch ist dies für große Systeme fast immer der Fall – höhere Energie bedeutet mehr Möglichkeiten, diese Energie unter den mikroskopischen Freiheitsgraden zu verteilen.
Druck ist definiert als:
Der Druck entsteht dadurch, dass die Vergrößerung des Volumens mehr Mikrozustände zugänglich macht – insbesondere wenn Teilchen sich frei bewegen können. Daher nimmt die Entropie mit dem Volumen zu, und diese Zunahme (skaliert mit der Temperatur) definiert den vom System ausgeübten Druck. Negativer Druck ist jedoch kein Zeichen thermodynamischer Instabilität.
Wenn die Teilchenzahl ebenfalls als thermodynamische Variable behandelt wird, ist das chemische Potential definiert als:
Dies drückt aus, wie sich die Entropie ändert, wenn ein Teilchen zum System hinzugefügt wird, bei konstanter Energie und konstantem Volumen. Es ist besonders wichtig bei der Untersuchung von Systemen, die Teilchen austauschen können (großkanonisches Ensemble), aber die Definition bleibt auch im mikrokanonischen Ensemble als formale Identität gültig.
Zusammenfassend können wir diese Definitionen neu zuweisen:
erneut zuweisen:
Dies ist tatsächlich der 1. Hauptsatz der Thermodynamik.
Und es ist einfach, alle Dinge in diesem Abschnitt auf Mehrkomponentensysteme zu erweitern. Erweitern Sie einfach zu
Nehmen wir als Beispiel das ideale Gas im mikrokanonischen Ensemble:
Betrachten Sie ein klassisches ideales Gas aus ununterscheidbaren Teilchen der Masse , eingeschlossen in das Volumen . Die Hamilton-Funktion ist rein kinetisch:
Die Oberflächenfläche ist definiert als:
Integration über die Koordinaten (ergibt ):
Setzen Sie . Verwenden Sie die Oberflächenformel einer -dimensionalen Kugel:
Für :
Wenden Sie die Stirling-Approximation für großes an:
Behalten Sie extensive Terme bei:
Dies ist die Sackur-Tetrode-Entropie.
Skalieren Sie Variablen mit :
Temperatur:
Druck:
Chemisches Potential:
2.4.2 Kanonisches Ensemble
Im Gegensatz zum mikrokanonischen Ensemble beschreibt das kanonische Ensemble Systeme im thermischen Gleichgewicht mit einem Wärmebad bei fester Temperatur. Es erlaubt Variationen von , was im mikrokanonischen Ensemble fest ist.
Betrachten Sie ein großes, isoliertes System mit der Gesamtenergie (mikrokanonisches Ensemble). Wenn wir das gesamte System in 2 Teile teilen: Teilsystem und Reservoir, wird die Energie in 3 Teile geteilt:
System: Kleines Teilsystem mit Hamilton-Funktion
Wärmebad: Großes Reservoir mit Hamilton-Funktion
Wechselwirkung: Wechselwirkung zwischen dem System und dem Reservoir
Bei großen Systemen ist die Wechselwirkung jedoch extrem schwach im Vergleich zu internen Wechselwirkungen. Daher wird die Wechselwirkungs-Hamilton-Funktion vernachlässigt.
Das System und das Reservoir tauschen Energie durch eine diatherme Wand aus, mit festem und für beide.
System des kanonischen Ensembles
Nach Boltzmanns Postulat hat jeder Mikrozustand des kombinierten Systems gleiche Wahrscheinlichkeit. Die Gesamtzahl der Mikrozustände ist:
Wir teilen dieses Integral auf, indem wir einfügen:
Betrachten Sie nun den Mikrozustand im Teilsystem. Gegeben , ist fest, aber variiert noch. Es gibt viele ‘s, die die Energie von haben. Daher ist die Wahrscheinlichkeitsdichte, dass das System im Mikrozustand ist, proportional zur Anzahl der verfügbaren Badzustände:
wobei die Boltzmann-Entropie des Bades ist.
Da das Bad viel größer als das System ist (), erweitern wir:
Definieren Sie die inverse Temperatur:
Somit:
Die Proportionalitätskonstante definiert die Zustandssumme:
ergibt die kanonische Verteilung:
Die Wahrscheinlichkeitsdichte, dass das System die Energie hat, ist:
wobei die Zustandsdichte des Systems ist.
Die Zustandssumme, , ist eine äußerst wichtige Funktion in der statistischen Mechanik. (Die Zustandssumme des mikrokanonischen Ensembles ist ). Sie dient als Erzeugungsfunktion für alle thermodynamischen Eigenschaften des Systems.
Definieren wir zuerst die freie Energie:
wird als Helmholtz-Freie Energie bezeichnet. Durch die Helmholtz-Freie Energie und die Zustandssumme können wir fast alle thermodynamischen Größen berechnen.
Mittlere Energie:
Gibbs-Shannon-Entropie:
Dieses Ergebnis kann die Beziehung zwischen der Helmholtz-Freien Energie und der Energie ableiten:
Ableiten und das Ergebnis für einsetzen, erhalten wir:
Energievarianz:
Wir haben auch:
Daher:
Dies zeigt die Äquivalenz zum mikrokanonischen Ensemble im thermodynamischen Limes.
Die konstante Temperatur des Bades ergibt sich aus seiner Größe
Seltene hochenergetische Zustände werden exponentiell durch unterdrückt
Freie Energie kodiert den Wettbewerb zwischen Energie und Entropie
2.4.3 Großkanonisches Ensemble
Das großkanonische Ensemble ist fast dasselbe wie das kanonische Ensemble, außer dass ebenfalls zwischen dem Teilsystem und dem Reservoir austauschbar ist.
System des großkanonischen Ensembles
Stellen Sie sich ein großes, isoliertes System vor, das durch seine Gesamtenergie , die gesamte Teilchenzahl und das gesamte Volumen charakterisiert ist. Dieses übergeordnete System kann durch seine mikrokanonische Zustandssumme beschrieben werden, , wobei die gesamte Hamilton-Funktion und die Integration über alle möglichen Mikrozustände bedeutet.
Nun betrachten wir die Aufteilung dieses isolierten Systems in ein kleines Teilsystem (bezeichnet mit dem Index ‘s’) und ein riesiges Reservoir (Index ‘r’). Die Gesamtzahl der Teilchen ist erhalten, was bedeutet . Die mikrokanonische Zustandssumme kann dann ausgedrückt werden als eine Summe über mögliche Teilchenzahlen im Teilsystem () und ein Integral über mögliche Energien im Teilsystem ():
Die Wahrscheinlichkeitsdichte, das Teilsystem mit Energie und Teilchenzahl zu finden, ist gegeben durch:
Hier ist die Entropie des Reservoirs. Für ein ausreichend großes Reservoir können wir eine Taylor-Entwicklung seiner Entropie um die Gesamtenergie und Teilchenzahl des isolierten Systems durchführen:
In dieser Entwicklung ist die inverse Temperatur (wobei die Temperatur ist) und das chemische Potential. Wenn wir dies zurück in die Wahrscheinlichkeitsdichte-Gleichung einsetzen, gelangen wir zu einem entscheidenden Ausdruck für die Wahrscheinlichkeitsdichte im großkanonischen Ensemble:
Der Nenner, , ist bekannt als die großkanonische Zustandssumme:
Diese Zustandssumme normalisiert effektiv die Wahrscheinlichkeit. Die Wahrscheinlichkeit eines bestimmten Mikrozustands () des Teilsystems mit Teilchen ist dann:
Die Normierungsbedingung erlaubt uns, die großkanonische Zustandssumme in Bezug auf die Hamilton-Funktion für ein System mit Teilchen auszudrücken:
Ein bemerkenswerter Aspekt des großkanonischen Ensembles ist seine direkte Beziehung zum kanonischen Ensemble. Die kanonische Zustandssumme beschreibt ein System mit einer festen Teilchenzahl , einem festen Volumen und einer festen Temperatur . Die großkanonische Zustandssumme kann als eine Summe über alle möglichen kanonischen Ensembles betrachtet werden, gewichtet mit dem Faktor :
Diese Gleichung zeigt elegant, dass das großkanonische Ensemble im Wesentlichen eine statistische Verschmelzung vieler kanonischer Ensembles ist, die jeweils einer anderen Teilchenzahl entsprechen. Diese Formulierung ist besonders leistungsfähig für Systeme, bei denen die Teilchenzahl keine erhaltene Größe ist.
So wie die Helmholtz-Freie Energie aus der kanonischen Zustandssumme abgeleitet wird, wird das großkanonische Potential, , aus der großkanonischen Zustandssumme definiert:
Das großkanonische Potential bietet eine direkte Verbindung zu den thermodynamischen Eigenschaften des Systems. Aus der Gibbs-Shannon-Entropie können wir eine fundamentale Beziehung ableiten:
Durch Umstellen dieser Gleichung finden wir einen wichtigen Ausdruck für das großkanonische Potential in Bezug auf andere thermodynamische Größen:
Hier ist die Helmholtz-Freie Energie (). Die Ableitung des großkanonischen Potentials liefert mehrere entscheidende thermodynamische Beziehungen:
Aus diesem Differential können wir die folgenden thermodynamischen Beziehungen ableiten:
Mittlere Teilchenzahl:
Druck:
Mittlere Energie:
Aus dem ersten Hauptsatz der Thermodynamik und der Extensivität der Energie () wissen wir, dass . Die Kombination mit der Definition des großkanonischen Potentials führt zu einer bemerkenswerten Identität:
Diese Gleichung stellt eine direkte Verbindung zwischen dem großkanonischen Potential und dem Druck-Volumen-Produkt des Systems her. Darüber hinaus kann die Gibbs-Freie Energie gleich gezeigt werden:
Dies impliziert, dass das chemische Potential die Gibbs-Freie Energie pro Teilchen darstellt, . Die Ableitung der Gibbs-Freien Energie führt zur Gibbs-Duhem-Relation:
Diese Relation hebt die gegenseitige Abhängigkeit intensiver Variablen (Temperatur, Druck und chemisches Potential) hervor. Auf Teilchenbasis kann sie geschrieben werden als:
wobei die Entropie pro Teilchen und das Volumen pro Teilchen ist.
Ein Schlüsselaspekt des großkanonischen Ensembles ist seine Fähigkeit, Fluktuationen in Energie und Teilchenzahl zu beschreiben, die aufgrund der Kopplung des Systems an das Reservoir inhärent zulässig sind. Die mittleren quadratischen Fluktuationen sind definiert als:
Diese Fluktuationen sind direkt mit Ableitungen der großkanonischen Zustandssumme verbunden:
Fluktuationen der Teilchenzahl:
Kreuzfluktuationen von Energie und Teilchenzahl:
Für makroskopische Systeme skalieren diese Fluktuationen als und sind daher im Vergleich zu den mittleren Werten, die als skalieren, vernachlässigbar. Das bedeutet, dass für große Systeme die Vorhersagen des großkanonischen Ensembles für mittlere Größen mit denen anderer Ensembles übereinstimmen werden. Das großkanonische Ensemble bleibt jedoch unschätzbar wertvoll für das Verständnis und die Berechnung von Eigenschaften offener Systeme, bei denen der Teilchenaustausch entscheidend ist.
2.4.4 Zusammenfassung
Im thermodynamischen Limes, wo die Teilchenzahl und das Volumen gegen unendlich gehen, während die Dichte konstant bleibt, liefern das mikrokanonische (NVE), kanonische (NVT) und großkanonische (µVT) Ensemble identische thermodynamische Vorhersagen. Diese Äquivalenz beruht auf der Unterdrückung relativer Fluktuationen makroskopischer Variablen. Innerhalb des kanonischen Ensembles nehmen Energiefluktuationen mit zunehmender Systemgröße ab, wobei relative Energiefluktuationen als skalieren. Wenn , verschwinden diese Fluktuationen, wodurch die scharf ausgeprägte Energieverteilung von einer Beschreibung mit fester Energie im mikrokanonischen Ensemble nicht zu unterscheiden ist.
Ähnlich werden im großkanonischen Ensemble sowohl Teilchenzahl- als auch Energiefluktuationen vernachlässigbar. Teilchenzahlfluktuationen gehorchen , was im thermodynamischen Limes verschwindet und die Teilchenzahl effektiv fixiert. Gleichzeitig verschwinden Energiefluktuationen relativ zur mittleren Energie, was das Ensemble mit den kanonischen und mikrokanonischen Rahmenwerken in Einklang bringt. Die thermodynamischen Potentiale jedes Ensembles – Entropie , Helmholtz-Freie Energie und großkanonisches Potential – sind durch Legendre-Transformationen streng miteinander verbunden. Diese Transformationen werden im thermodynamischen Limes exakt, was identische Vorhersagen für intensive Größen wie Druck oder Energiedichte über alle Ensembles hinweg sicherstellt.
Somit etablieren die verschwindenden relativen Fluktuationen und die mathematische Konsistenz der thermodynamischen Potentiale die Äquivalenz der drei Ensembles für makroskopische Systeme.
2.5 Extremumprinzipien
2.5.1 Thermodynamische Potentiale
Wir kennen bereits die Energie , die Helmholtz-Freie Energie und das großkanonische Potential .
Führen Sie eine Legendre-Transformation durch:
Dies wird als Gibbs-Freie Energie definiert.
Nehmen Sie eine Differentialform, wir erhalten:
Wir können auch die Enthalpie definieren:
Was wir klären müssen ist, dass die Differentialvariablen in der Differentialgleichung die natürlichen Variablen dieses Potentials sind.
Nehmen Sie die Energie als Beispiel: wir haben
und wir können dieses Beispiel auf erweitern. Zum Beispiel können wir sagen:
2.5.2 Maxwell-Relationen
Die thermodynamischen Potentiale sind wohldefinierte, eindeutige Funktionen ihrer natürlichen Variablen. Bei gemischten zweiten Ableitungen wie kann die Reihenfolge der partiellen Ableitung vertauscht werden:
da und , erhalten wir:
Dies führt zu einer sehr großen Anzahl von Identitäten zwischen partiellen Ableitungen verschiedener thermodynamischer Variablen, die alle Maxwell-Relationen genannt werden: Da diese Differentiale exakt sind, ergeben gemischte partielle Ableitungen Maxwell-Relationen. Wir listen alle 15 Maxwell-Relationen unten auf:
Wie bereits in Lektion 2 gezeigt, maximiert der Gleichgewichtszustand eines isolierten Systems die Gibbs-Shannon-Entropie unter den Nebenbedingungen der Wahrscheinlichkeitsnormierung und der festen Energie (was bedeutet, dass die Wahrscheinlichkeitsdichtefunktion außerhalb der Energieschale verschwindet). Die entropie-maximierende Wahrscheinlichkeitsverteilung ist eine Gleichwahrscheinlichkeitsverteilung auf der Energieschale. Daher ist das Prinzip der maximalen Entropie letztlich äquivalent zu Boltzmanns Postulat der gleichen Wahrscheinlichkeit. Entweder kann als Ausgangspunkt der statistischen Gleichgewichtsmechanik verwendet werden.
Betrachten Sie ein System mit kontinuierlichen Mikrozuständen in thermischem Kontakt mit einem Wärmebad bei Temperatur , mit festem Volumen und Teilchenzahl . Sei die Energie des Mikrozustands und die Wahrscheinlichkeitsdichte eines beliebigen Nichtgleichgewichtszustands. Die mittlere Energie ist:
und die Gibbs-Shannon-Entropie ist:
Definieren Sie die Nichtgleichgewichts-Helmholtz-Freie Energie als:
wobei . Für festes suchen wir das , das extremisiert, unter der Nebenbedingung der Wahrscheinlichkeitsnormierung:
Einführung eines Lagrange-Multiplikators , das zu extremisierende Funktional ist:
Die erste Variation, , ergibt:
was eine kanonische Verteilung ergibt:
Die zweite Variation, , bestätigt, dass dies ein Minimum ist. Diese entspricht der kanonischen Verteilung, abgeleitet über Boltzmanns Postulat. Somit erhalten wir das folgende Extremumprinzip für den Gleichgewichtszustand eines thermisch offenen Systems, das Energie mit einem Gleichgewichts-Wärmebad austauschen kann:
Im Gleichgewicht wird die Nichtgleichgewichts-Helmholtz-Freie Energie eines thermisch offenen Systems minimiert.
Beachten Sie, dass dieses Prinzip sowohl für kleine als auch für große Systeme gültig ist. Es ist einfach zu überprüfen, dass dieses Prinzip auch in die folgende äquivalente Form umformuliert werden kann:
Im Gleichgewicht wird die Gibbs-Shannon-Entropie eines thermisch offenen Systems maximiert unter den Nebenbedingungen der Normierung und der festen mittleren Energie.
Betrachten Sie nun ein System, das Energie und Teilchen mit einem Bad bei Temperatur und chemischem Potential austauscht, bei festem Volumen . Sei die diskrete Teilchenzahl und ein kontinuierlicher Mikrozustand für gegebenes mit Energie . Für einen Nichtgleichgewichtszustand ergeben sich aus der Wahrscheinlichkeitsdichte die Mittelwerte:
und die Gibbs-Shannon-Entropie:
Das Nichtgleichgewichts-großkanonische Potential ist definiert als:
Wir suchen , das extremisiert, unter der Nebenbedingung der Normierung:
Unter Verwendung eines Lagrange-Multiplikators ist das Funktional:
Setzen der Funktionalableitung auf Null:
finden wir die großkanonische Verteilung:
wobei das chemische Potential ist und die großkanonische Zustandssumme ist. Wir können auch zeigen, dass die zweite Variation positiv ist: , was bestätigt, dass dies ein Minimum des großkanonischen Potentials ist. Dies etabliert das folgende Extremumprinzip für ein offenes Gleichgewichtssystem, das Energie und Teilchen mit seiner Umgebung austauscht:
Im Gleichgewicht wird das Nichtgleichgewichts-großkanonische Potential eines offenen Systems minimiert.
Eine äquivalente Darstellung dieses Extremumprinzips ist die folgende:
Im thermodynamischen Gleichgewicht wird die Gibbs-Shannon-Entropie eines offenen Systems maximiert unter den Nebenbedingungen der Wahrscheinlichkeitsnormierung und der festen mittleren Energie sowie der festen mittleren Teilchenzahl.
2.6 Gleichgewichtsbedingungen
2.6.1 Thermisches Gleichgewicht
Betrachten Sie ein isoliertes System, das aus zwei Teilsystemen besteht, die Energie austauschen können (dies ist tatsächlich das kanonische Ensemble). Die Anzahl der zugänglichen Mikrozustände für eine gegebene Energieaufteilung ist:
und die entsprechende Wahrscheinlichkeit ist
Gemäß dem Prinzip der maximalen Entropie müssen wir maximieren. Um den Wert von zu finden, der maximiert (bezeichnet als ), sollten wir die Ableitung nehmen:
Dies ergibt
Erweiterung von um , mit
ergibt eine Gaußsche Verteilung
und die Fluktuation
Wenn , dann ist die Entropieänderung, die mit einer kleinen Energieübertragung von System 1 zu System 2 verbunden ist:
Dies deutet darauf hin, dass die Entropie zunimmt, wenn Energie vom heißeren zum kälteren Teilsystem fließt.
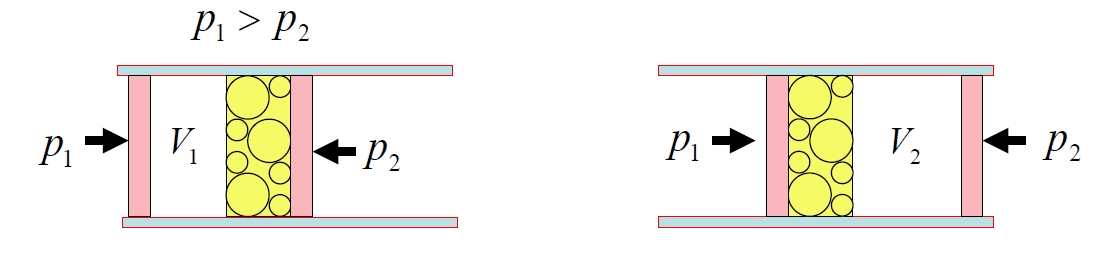
2.6.2 Mechanisches Gleichgewicht
Betrachten Sie nun zwei Teilsysteme, die Volumen durch einen beweglichen, reibungsfreien Kolben austauschen können, während die Gesamtenergie und das Gesamtvolumen konstant bleiben. Sei Teilsystem 1 das Volumen einnehmen und Teilsystem 2 . Die Gesamtentropie ist:
Die wahrscheinlichste Konfiguration maximiert die Gesamtentropie bezüglich Variationen von :
da , haben wir:
Unter der Annahme, dass das thermische Gleichgewicht bereits hergestellt ist, vereinfacht sich dies zu:
Dies ist das mechanische Gleichgewicht.
Ähnlich wie bei der Temperatur können wir eine solche Beziehung ableiten:
Dies stimmt mit der alltäglichen Vorstellung von Druck als einer Kraft überein, die eine Expansion antreibt. Im Gleichgewicht verschwindet die treibende Kraft; außerhalb des Gleichgewichts führt sie zu gerichteter Bewegung, die die Entropie erhöht. Daher ist die übliche physikalische Interpretation von Druck als expansiver Kraft vollständig konsistent mit seinen thermodynamischen und statistischen Definitionen.
2.6.3 Chemisches Gleichgewicht
Ebenso können wir auch das chemische Gleichgewicht ableiten. Betrachten Sie ein System mit mehreren Komponenten, wir haben:
Unter Verwendung der Identität:
Wir haben:
Daher fließen Teilchen spontan von höherem zu niedrigerem chemischem Potential, genauso wie Wärme von heiß nach kalt fließt und Volumen von hohem zu niedrigem Druck expandiert. Das chemische Potential kann somit als eine verallgemeinerte “Kraft” interpretiert werden, die den Teilchenaustausch in Richtung Gleichgewicht antreibt.
2.7 Stabilitätsbedingung
2.7.1 Entropiestabilität
Betrachten Sie zwei Systeme mit denselben Makrozuständen (d. h. demselben ). Gemäß der Skalierungseigenschaft haben wir:
Wenden Sie nun kleine Störungen auf das System an:
Unser Ziel ist es, die Stabilitätsbedingung zu finden. Erinnern Sie sich, dass Stabilität der zweiten Ableitung entspricht. Also erweitern wir:
Das Differential der Entropie für ein einzelnes Teilsystem ist:
Wir können diese Gleichungen differenzieren. Schließlich erhalten wir:
Da die Entropie maximiert wird, wenn wir wollen, dass das System stabil ist, . Daher:
Dann:
2.7.2 Diagonalisierung von
Wir können die zweite Variation der Entropie als quadratische Form ausdrücken:
wobei , , usw.
Aus der thermodynamischen Identität:
können wir nach auflösen:
Einsetzen in eliminiert Kreuzterme mit :
Um zu vereinfachen, definieren wir neue Koeffizienten:
und führen eine neue Variable ein:
Angenommen , dann:
dS =
Wir können eine Jacobi-Determinante ableiten:
Variablen ändern zu:
, ,
Da
dann können wir feststellen, dass
Einsetzen der Ergebnisse in :
Dies erfordert zwei unabhängige Stabilitätsbedingungen:
2.7.3 Chemische Stabilität
Wir betrachten . Dann reduziert sich die zweite Ableitung zu:
Dies führt zu einer weiteren Stabilitätsbedingung:
Für ein extensives System ist die Helmholtz-Freie Energie:
Chemisches Potential:
Differenzieren Sie nach :
Druckrelation:
Einsetzen:
Also führt chemische Stabilität zu .
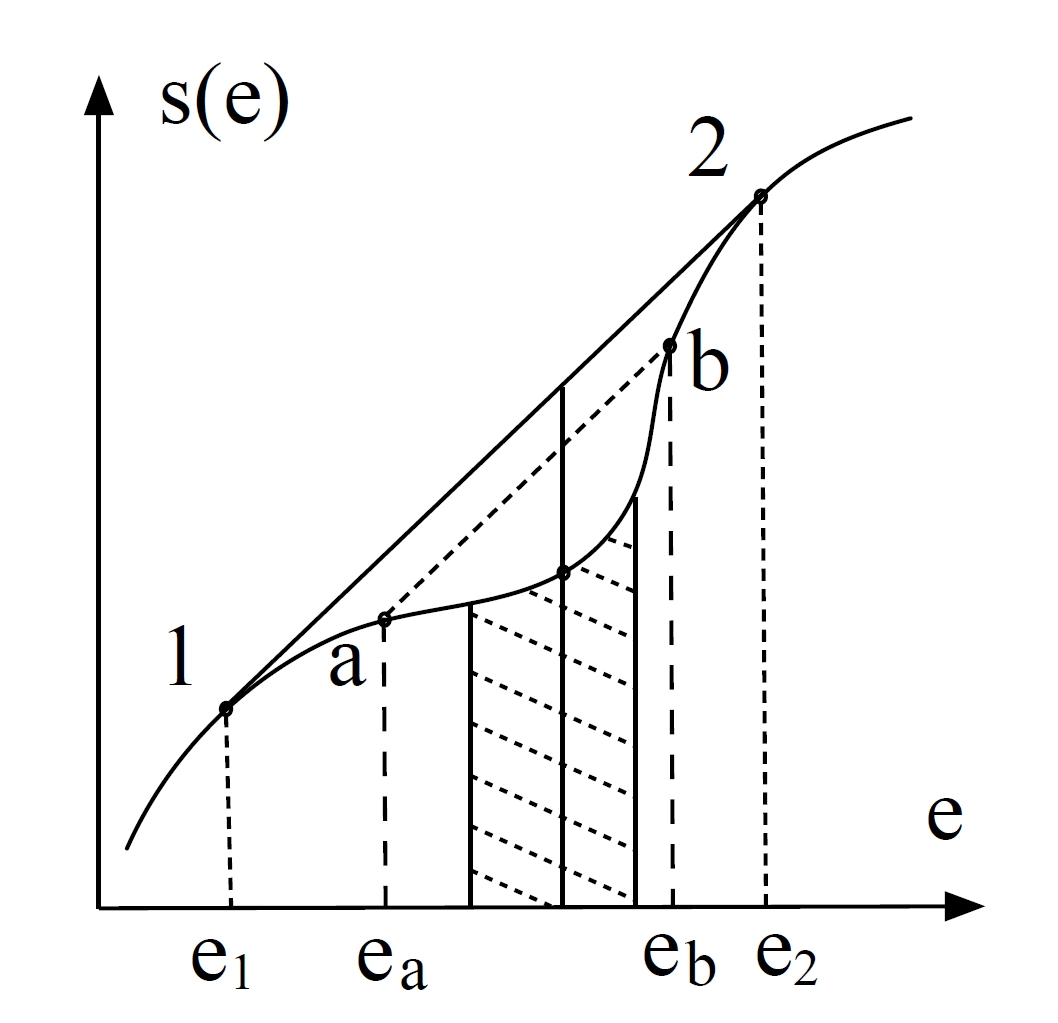
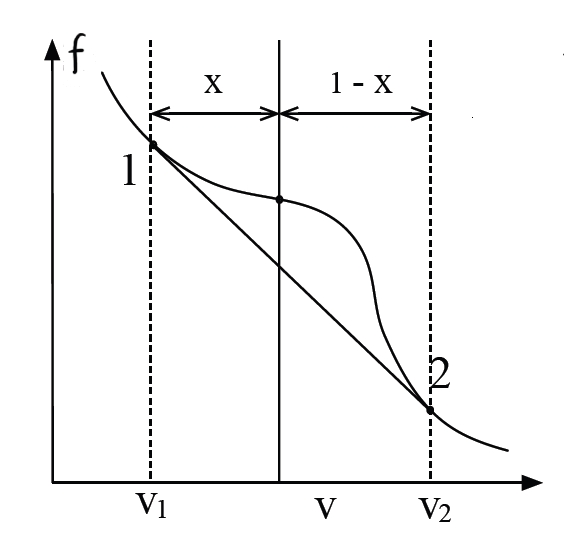
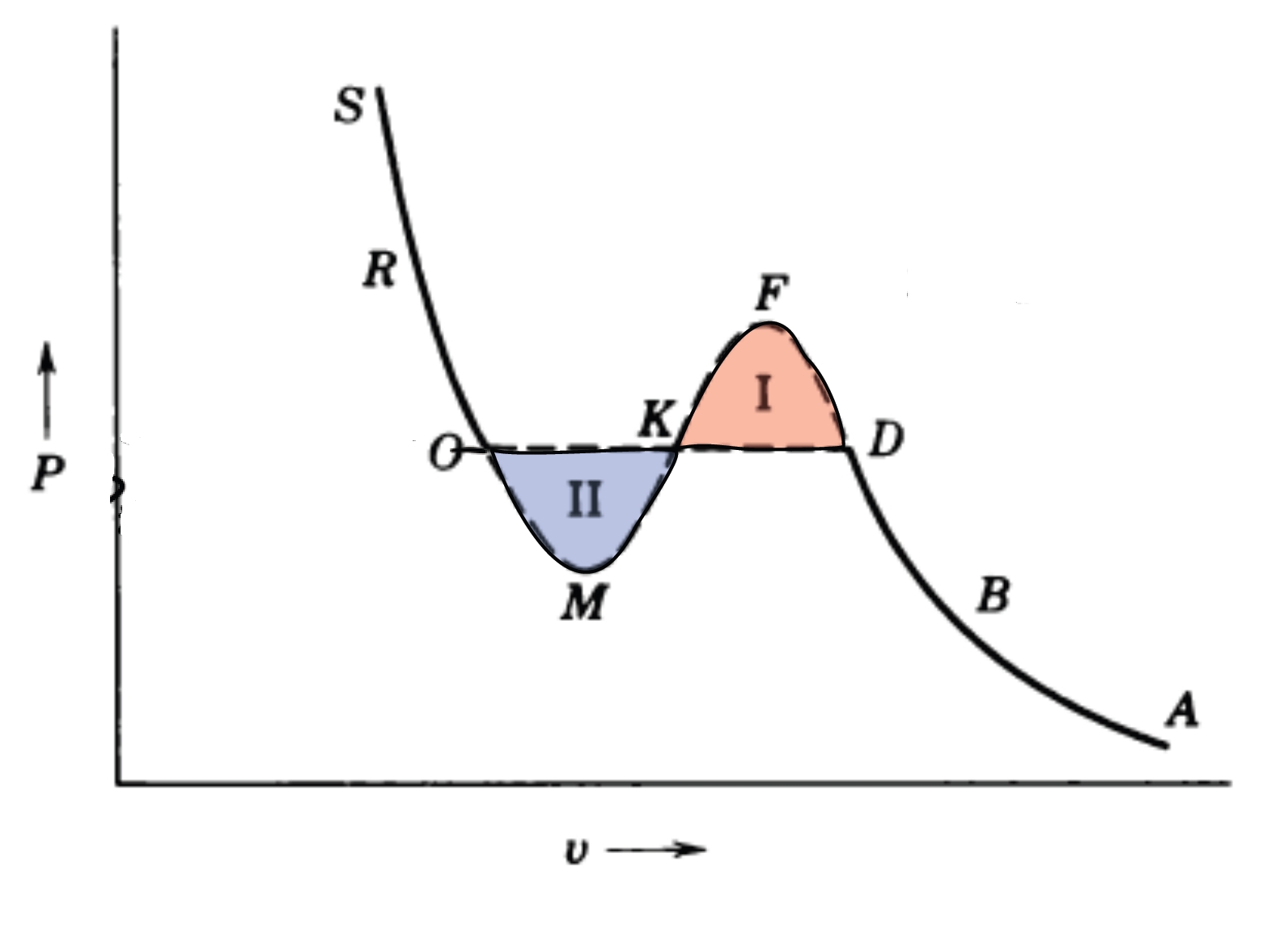
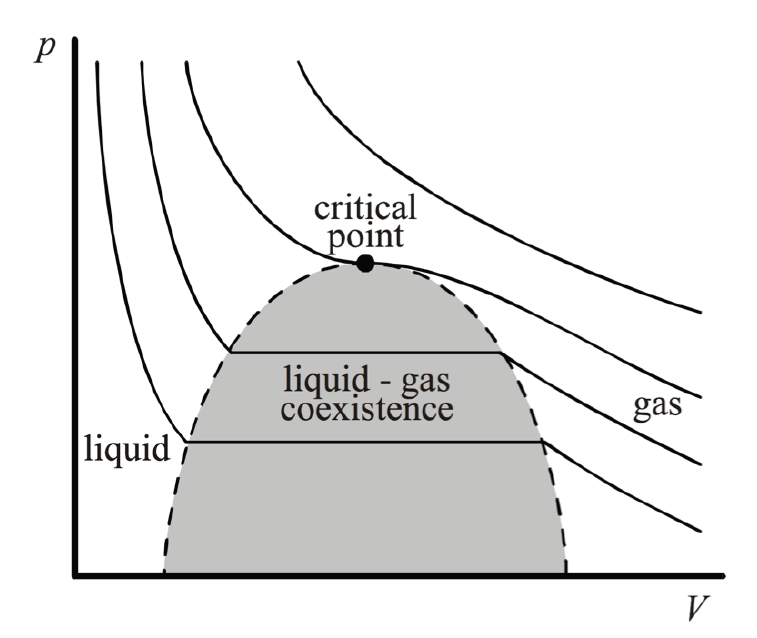
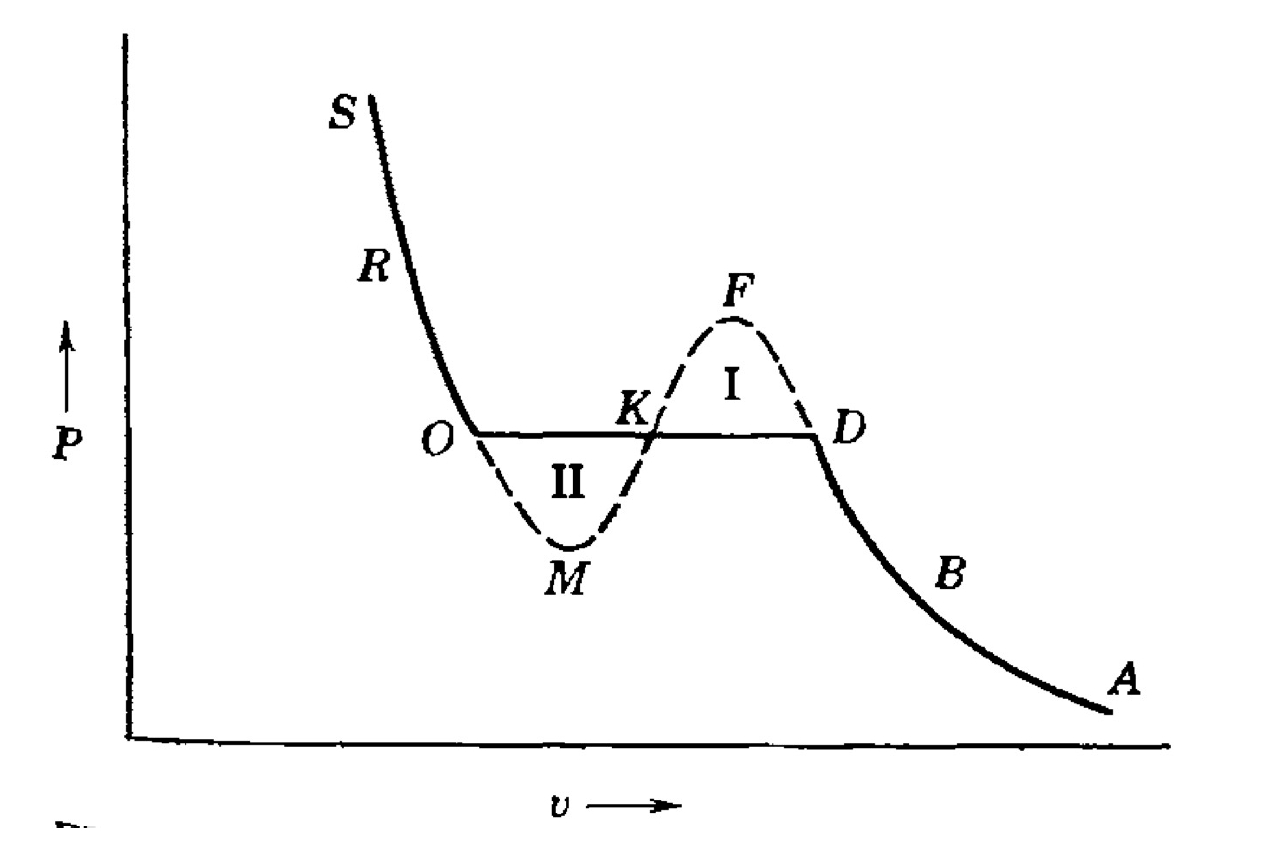
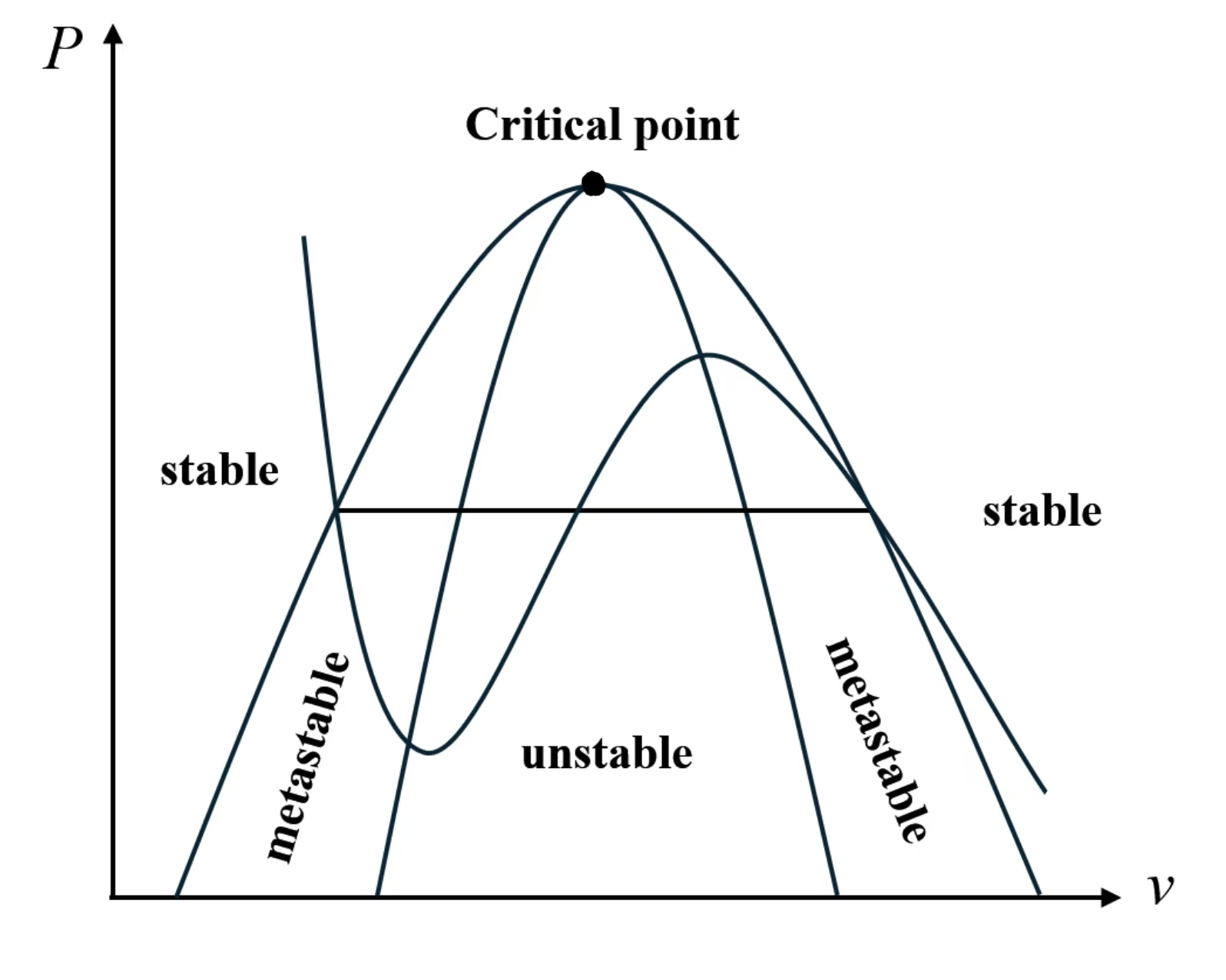
2.8 Phasenübergang erster Ordnung
2.8.1 Konflikt am Übergangspunkt
Phasenübergänge erster Ordnung, wie Sieden oder Schmelzen, sind gekennzeichnet durch Diskontinuitäten in thermodynamischen Observablen – wie Energie oder Volumen –, wenn externe Parameter wie Temperatur oder Druck variiert werden. Diese Diskontinuitäten signalisieren die Koexistenz makroskopisch unterschiedlicher Phasen und entstehen aus einem subtilen Versagen der Stabilität in thermodynamischen Potentialen. Ihr Verständnis erfordert eine präzise Analyse der Entropiekrümmung (der Ableitung zweiter Ordnung), der Ensemble-Äquivalenz und der Geometrie thermodynamischer Funktionen.
Betrachten Sie zunächst das mikrokanonische Ensemble. Wir nehmen an, das Volumen ist fest, sodass wir uns nicht um und kümmern müssen. Die Temperatur ergibt sich:
Wir können die Wärmekapazität finden:
Wir verwenden die Superskript MC, um anzuzeigen, dass sie mit dem mikrokanonischen Ensemble berechnet wurde.
Normalerweise ist konkav, sodass , was zu einer positiven Kapazität führt. Aber für einige Systeme kann konvex sein, was macht. Sie können es in der folgenden Abbildung deutlich sehen:
Entropiekurve am Phasenübergangspunkt
Nun untersuchen wir dasselbe System mit dem kanonischen Ensemble. Aufgrund der Äquivalenz verschiedener Ensembles im thermodynamischen Limes sollte das kanonische Ensemble dasselbe Ergebnis wie das mikrokanonische Ensemble liefern.
Wir haben die Zustandssumme:
und die Helmholtz-Freie Energie:
Die mittlere Energie ist:
Wir bezeichnen die Kapazität im kanonischen Ensemble als . Sie kann berechnet werden durch:
Fluktuationen sind immer positiv, sodass auch jederzeit positiv sein muss. Dies führt jedoch zu einem Konflikt, wenn die Entropie konvex ist. Ein solch seltsamer Konflikt muss ein ungewöhnliches physikalisches Phänomen beinhalten, dies ist der Phasenübergang erster Ordnung.
2.8.2 Phasenübergang im mikrokanonischen Ensemble
Um den Konflikt zwischen den Ensembles zu lösen, verwenden wir einen geometrischen Ansatz, der den physikalischen Mechanismus der Phasentrennung aufdeckt.
Kehren wir zur Entropiekurve zurück.
Entropiekurve am Phasenübergangspunkt
Für jede Energie zwischen und können wir eine höhere Entropie durch Bildung einer Mischung aus zwei Phasen erreichen:
Anteil in Phase (Energie , Entropie )
Anteil in Phase (Energie , Entropie )
Die Mischungsentropie übersteigt die homogene Entropie:
Die optimalen Phasen (, ) werden durch die Doppeltangentenbedingung bestimmt:
Die gerade Linie, die und verbindet, muss an beiden Punkten tangential zu sein.
Dies erfordert gleiche Steigungen bei und :
Die Gleichheit der Steigungen impliziert:
Dies garantiert thermisches Gleichgewicht zwischen den beiden Phasen. Die Temperatur an diesem Punkt wird als bezeichnet und kritische Temperatur genannt. Darüber hinaus können wir chemisches Gleichgewicht zeigen:
Die Gleichgewichtsentropie folgt der konkaven Hülle:
wobei die Mischungsentropie die lineare Interpolation ist:
Zwischen und ist . Daher können wir die Kapazitätsdivergenz finden:
Der scheinbare Widerspruch zwischen der mikrokanonischen Wärmekapazität und der kanonischen Wärmekapazität wird durch eine fundamentale Singularität aufgelöst. Diese Divergenz eliminiert die Unterscheidung zwischen positiven und negativen Werten – die Polsingularität macht die Vorzeichenambiguität physikalisch bedeutungslos. Die “negative” im mikrokanonischen Ensemble und die strikt positive Anforderung im kanonischen Ensemble konvergieren zu identischem divergentem Verhalten am Phasenübergang.
Dies stimmt mit unseren Lebenserfahrungen überein. Wir wissen, dass Schmelzen und Sieden zusätzliche Wärme absorbieren, während die Temperatur unverändert bleibt. Gemäß der Definition der Kapazität ist sie zwangsläufig unendlich. Die während des Prozesses stattfindende Wärmeänderung wird als latente Wärme bezeichnet. Wir werden weitere Details in den folgenden Abschnitten klären.
Wir müssen darauf achten, dass die konvexe Kurve ein Ergebnis der Zustandsgleichung ist und die Doppeltangentenlinie zeigt, wie sich die Entropie in der Realität entwickelt. Der konvexe Bereich der Entropiekurve ergibt sich direkt aus der fundamentalen Zustandsgleichung des Systems – dieser gekrümmte Pfad repräsentiert alle möglichen homogenen Konfigurationen, die durch mikroskopische Wechselwirkungen bestimmt werden. Dieses konvexe Segment bleibt jedoch aufgrund seiner Instabilität in physikalischen Systemen thermodynamisch verboten. Die Doppeltangentenlinie zeigt, was tatsächlich passiert: Systeme durchlaufen spontan eine Phasentrennung, um diesem geradlinigen Pfad in der Entropie-Energie-Ebene zu folgen.
Diese geometrische Dualität – gekrümmte Zustandsgleichung versus geradliniger Phasenübergangspfad – erklärt, warum kochendes Wasser bei 100°C Wärme absorbiert, ohne die Temperatur zu ändern: Das System entwickelt sich entlang der Doppeltangentenlinie und nicht entlang des instabilen gekrümmten Entropiepades, der für homogene Zustände vorhergesagt wird.
2.8.3 Phasenübergang im kanonischen Ensemble
Da der Konflikt gelöst wurde, werden wir das gesamte System mit dem kanonischen Ensemble ableiten.
Betrachten Sie ein einfaches Fluid (-System) mit Teilchenzahl und Volumen in Kontakt mit einem Wärmebad bei Temperatur . Gemäß dem Prinzip der minimalen freien Energie wird die Helmholtz-Freie Energie im Gleichgewicht minimiert. Angenommen, wir berechnen die Helmholtz-Freie Energie pro Teilchen , unter Annahme von Homogenität. Die Differentialform ist:
Der Druck wird durch Ableitung erhalten:
Die mechanische Stabilitätsbedingung erfordert:
Wenn diese Bedingung in einem Intervall verletzt wird, tritt Phasentrennung auf.
Helmholtz-Freie Energie am Phasenübergangspunkt
Mit derselben Methode finden wir die beiden Punkte des Phasenübergangs über . Übergangspunkte erfüllen:
wobei der Koexistenzdruck ist. Die Doppeltangente ist:
Für minimiert das System die freie Energie durch Trennung in Phasen:
Die Gleichgewichts-freie Energie ist die konvexe Hülle:
Wir können immer noch Gleichgewichtsbedingungen ableiten:
Thermisches Gleichgewicht: Gleiche
Mechanisches Gleichgewicht:
Chemisches Gleichgewicht:
2.8.4 Maxwell-Konstruktion
Die Helmholtz-Freie Energie-Kurve zeigt einen konkaven Bereich innerhalb des Phasenübergangsintervalls. Diese Konkavität verletzt die mechanische Stabilitätsbedingung, die erfordert, und zeigt an, dass homogene Zustände in diesem Bereich thermodynamisch instabil sind.
Die Bedingung für chemisches Gleichgewicht für koexistierende Phasen ist gegeben durch:
Unter Verwendung der Druckdefinition:
werten wir das Integral aus:
Dieses Ergebnis ist die Maxwell-Konstruktion, deren Essenz die Durchsetzung des Gleichgewichts des chemischen Potentials ist. Geometrisch erfordert sie, dass die Fläche von Region I gleich der von Region II in der folgenden Abbildung ist:
Maxwell-Konstruktion in der -Kurve
2.8.5 Latente Wärme
Während Phasenübergängen erster Ordnung wie Sieden oder Schmelzen absorbieren oder geben Systeme signifikante Energie ab, während sie eine konstante Temperatur beibehalten – ein Phänomen, das am vertrautesten ist, wenn Wasser bei 100°C kocht und bei dieser Temperatur bleibt, bis es vollständig verdampft ist. Diese verborgene Energieaustausch, latente Wärme (bezeichnet als ) genannt, bricht oder bildet molekulare Bindungen, anstatt die thermische Bewegung zu erhöhen.
Für ein System von Teilchen wird die gesamte latente Wärme quantifiziert als:
wobei den Entropieunterschied pro Teilchen zwischen den Phasen darstellt – zum Beispiel erfordert die Verdampfung von Wasser ungefähr , um intermolekulare Kräfte zu überwinden. Entscheidend ist, dass die latente Wärme äquivalent der Enthalpiedifferenz zwischen koexistierenden Phasen ist, ausgedrückt als , eine Beziehung, die sich direkt aus der Gleichheit der chemischen Potentiale () im Phasenübergleichgewicht ergibt.
Die Koexistenz von Phasen in der Temperatur-Druck-()-Ebene wird durch die Gleichheit der chemischen Potentiale definiert, die eine Schnittkurve zweier Oberflächen im -Raum beschreibt, wo Phasen gleich stabil sind; anderswo dominiert die Phase mit niedrigerem , da sie die Gibbs-Freie Energie minimiert. Wenn kleine Verschiebungen entlang dieser Koexistenzkurve () verfolgt werden, führt die Erhaltung des chemischen Gleichgewichts zur Clausius-Clapeyron-Gleichung:
wobei die spezifische Volumenänderung pro Teilchen ist. Diese Gleichung zeigt, wie sich die Koexistenzdrücke mit der Temperatur verschieben: Für die meisten Substanzen dehnt sich das Volumen beim Schmelzen oder Sieden aus (), was zu einer positiven Steigung () führt; Wasser zeigt jedoch ein anomales Verhalten, bei dem das Schmelzen von Eis das Volumen verringert (, da flüssiges Wasser dichter ist), was zu einer negativen Steigung () führt, die erklärt, warum erhöhter Druck Eis bei konstanter Temperatur schmilzt – ein Prinzip, das Schlittschuhlaufen ermöglicht. Für Wasser, das bei siedet, ergeben die Werte und , was eine erhebliche Druckempfindlichkeit nahe den Phasengrenzen anzeigt.
2.8.6 Stabil und Metastabil
Der kritische Punkt ist der thermodynamische Zustand, bei dem sich unterscheidbare flüssige und gasförmige Phasen nicht mehr unterscheiden lassen, gekennzeichnet durch:
Kritische Temperatur ()
Kritischer Druck ()
Kritisches Volumen ()
An diesem Punkt endet die Koexistenz von Flüssigkeit und Gas, die Oberflächenspannung verschwindet und thermodynamische Antwortfunktionen (z. B. Kompressibilität ) divergieren. Sie können es in der folgenden Abbildung deutlich sehen:
Kritischer Punkt
Nehmen wir das Van-der-Waals-Gas als Beispiel. Es hat die Zustandsgleichung:
wobei das Volumen pro Teilchen ist. Für Temperaturen unter ist die -Kurve nicht monoton, was bedeutet, dass es konkave Bereiche gibt. Dieser Bereich zeigt (Instabilität). Die Grenze des Phasenübergangs wird durch die Maxwell-Konstruktion konstruiert. Der kritische Punkt wird bestimmt durch:
Wenn wir dies für das Van-der-Waals-Gas lösen, ist das Ergebnis:
Wir müssen die Elemente in der -Abbildung veranschaulichen.
-Diagramm nach Maxwell-Konstruktion
Wir müssen diese Abbildung mit der Doppeltangentenlinie der Helmholtz-Freien Energie in Beziehung setzen. Die Punkte O und D sind die Grenzen des Phasenübergangs, die den Tangentenpunkten von entsprechen. Die Linie OD ist die Projektion der konstruierten Doppeltangentenlinie. Die Kurve OMFD ist die Projektion von vor der Konstruktion. Offensichtlich konvergieren mit steigender Temperatur die verdrehten Punkte M und F allmählich zu ihrem Mittelpunkt K und verschmelzen schließlich zu einem Punkt bei der kritischen Temperatur .
Was uns immer noch rätselhaft ist, ist die Bedeutung der Punkte M und F. Beachten Sie, dass zwischen M und F der instabile Bereich liegt, den wir oben erwähnt haben. M und F sind also die Grenzen der Instabilitätsbereiche. Die Segmente OM und FD entsprechen metastabilen Zuständen, in denen das System in einem lokalen Minimum der Helmholtz-Freien Energie verbleibt. Diese Zustände erfüllen lokale Stabilitätsbedingungen (), sind aber thermodynamisch dem getrennten Phasenzustand auf der Maxwell-Linie unterlegen. Klassische Beispiele sind:
Unterkühlte Flüssigkeit: Flüssigkeit bleibt unter (z. B. bis ) bestehen, da kinetische Barrieren die Eisnukleation behindern.
Überhitzte Flüssigkeit: Flüssigkeit existiert über (z. B. bis bei hohem Druck) ohne zu sieden.
Diese metastabilen Zustände sind empfindlich gegenüber Störungen:
Nukleation löst Kollaps aus: Einführung von Verunreinigungen, Vibrationen oder Dichteschwankungen treibt das System zum globalen Minimum – spontane Phasentrennung entlang der Maxwell-Linie OD.
Physikalische Mechanismus: Lokale Minima der freien Energie (OM/FD) sind durch Energieriegel von der Koexistenzlinie (OD) getrennt; sobald diese überwunden sind, setzt das System latente Wärme frei und erreicht das Gleichgewicht durch Phasentrennung.
Nun müssen wir die exakte Grenzlinie in der -Ebene berechnen. Wir werden eine Methode namens asymptotische Entwicklung verwenden. Nehmen wir immer noch das Van-der-Waals-Gas.
Um das Verhalten nahe dem kritischen Punkt zu analysieren, definieren wir reduzierte Variablen:
Die Entwicklung der Van-der-Waals-Gleichung um den kritischen Punkt ergibt:
Dies beschreibt die Flüssig-Gas-Koexistenz für .
Die Koexistenzvolumina (Flüssigkeit) und (Gas) erfüllen:
Gleicher Druck:
Gleiches chemisches Potential:
Maxwell-Flächenregel:
Nehmen wir asymptotische Entwicklungen an (Quadratwurzel, um den Polpunkt zu vermeiden):
Die Kombination der obigen Gleichungen und der Abgleich der Ordnungen von ergibt:
Anwendung der Flächenbedingung bestimmt :
Somit sind die Koexistenzvolumina:
und der Koexistenzdruck ist:
Die Ableitung bestimmt die Stabilität:
Die Spinodalpunkte (wo ) markieren die Stabilitätsgrenzen:
Hier ist eine Zusammenfassung:
Stabile Flüssigkeit:
Stabiles Gas:
Metastabile unterkühlte Flüssigkeit:
Metastabiler überhitzter Dampf:
Instabiler (spinodaler) Bereich:
Stabilitätsdiagramm für (horizontale Linie: Maxwell-Konstruktion)
2.8.7 Gibbssche Phasenregel
In diesem Abschnitt werden wir Mehrkomponentensysteme untersuchen. Für Mehrkomponentensysteme wird die Differentialgleichung der Energie erweitert zu:
Die Gibbs-Freie Energie ist hier besonders nützlich. Ihre Differentialform ist:
Diese Gleichung ergibt:
Die Vertauschbarkeit partieller Ableitungen führt dann zu:
Durch Inspektion der obigen Formeln können wir eine Einschränkung des chemischen Potentials finden:
Also:
Angenommen, wir haben ein System mit chemischen Komponenten und koexistierenden Phasen. Koexistierende Phasen bedeuten, dass die beiden Phasen eine makroskopische, beobachtbare Grenze haben. Zum Beispiel sind festes Eis, flüssiges Wasser, festes Alkohol drei koexistierende Phasen.
Wir nehmen an, dass Temperatur und Druck im gesamten System einheitlich und konstant sind,
wie es das Gleichgewicht erfordert.
Daher sind die einzigen verbleibenden intensiven
Variablen, die zur Charakterisierung jeder Phase benötigt werden, die relativen Zusammensetzungen. Wir bezeichnen als den Molenbruch der Komponente in Phase . Daher haben wir
Aufgrund dieser Einschränkung
hat jede Phase nur unabhängige Variablen.
Über alle Phasen hinweg
gibt es also unabhängige Variablen.
Während
neben den Brüchen auch das chemische Potential mit und zusammenhängt.
Daher hat einen Freiheitsgrad (DOF) von .
Als Nächstes erfordert das Gleichgewicht, dass alle chemischen Potentiale gleich sind:
Dies ergibt Gleichungen, die den DOF von reduzieren.
Dann ergibt sich der gesamte DOF zu
Dies wird als **Gibbssche Phasenregel** bezeichnet und gibt uns die Beziehung zwischen der Anzahl der Komponenten und der Anzahl der koexistierenden Phasen an.
Der DOF gibt an, wie viele Parameter angepasst werden können, während
der aktuelle Zustand (sowohl als auch ) unverändert bleibt.
Um die maximale Anzahl koexistierender Phasen zu bestimmen,
sollten wir auf 0 setzen und erhalten
Zum Beispiel haben wir eine Mischung aus Wasser und Alkohol.
Nach der Gibbsschen Phasenregel
können höchstens 4 Phasen koexistieren.
Sie könnten flüssiges Wasser, gasförmiges Wasser, flüssigen Alkohol, gasförmigen Alkohol sein.
Wenn wir die Temperatur senken, um Wasser einzufrieren,
wird gasförmiger Alkohol verschwinden.
2.8.8 Chemische Reaktionen
Betrachten Sie eine chemische Reaktion, an der Komponenten beteiligt sind:
wobei die -te chemische Spezies und ihr stöchiometrischer Koeffizient ist.
zeigt Produkte und für Reaktanten an.
Sei die Anzahl der Mol der Spezies .
Wir definieren die Reaktionskoordinate .
Sie ist gegeben durch
Die Molzahl jeder Spezies kann dann geschrieben werden als
wobei die anfängliche Menge der Spezies vor dem Reaktionsfortschritt ist.
Bei festem
wird die Gibbs-Energie geschrieben als
Differenzieren Sie beide Seiten, wir erhalten
Der zweite Term verschwindet, weil
So können wir die Gibbs-Energie mit der Gesamtänderung der Teilchenzahl darstellen:
wobei die Gibbs-Reaktionsenergie genannt wird.
Dies ist eine Größe, die den Reaktionsfortschritt anzeigt
(da während der Reaktion zunimmt).
Sie können sie als Ableitung von nach dem Reaktionsfortschritt betrachten.
In unserem direkten Sinne
ist , wenn die Reaktion das Gleichgewicht erreicht.
Wir werden dies als Nächstes beweisen, unter Verwendung des zweiten Hauptsatzes der Thermodynamik.
Betrachten Sie ein typisches kanonisches Ensemble-System, das aus einem Teilsystem und einem Reservoir besteht.
Die gesamte Entropieänderung darf nicht negativ sein:
Angenommen, das System wird gestört und die Makroparameter ändern sich um einen kleinen Wert.
wobei sich seine Energie und sein Volumen um und ändern.
Das Reservoir ändert sich entsprechend um und .
Dann kann ihre Entropieänderung geschrieben werden als
Da konstant sind, kann die gesamte Entropieänderung dann geschrieben werden als:
Dies ergibt .
Daher:
Dies impliziert, dass die Reaktion in der Richtung fortschreitet, die
reduziert.
Im Gleichgewicht gilt:
Somit minimiert das System seine Gibbs-Energie bei konstantem
und konstanten Gesamtatomzahlen.
Wenn , dann ist , was bedeutet, dass die Reaktion vorwärts geht,
während wenn , dann ist , was bedeutet, dass die Reaktion rückwärts geht.
In der Praxis deutet die stöchiometrische Gleichung darauf hin, dass die Reaktion
fortschreiten könnte, bis eine der Komponenten vollständig verbraucht ist.
Dies ist jedoch thermodynamisch nicht möglich.
Wenn die Molzahl einer beliebigen Spezies ,
divergiert das chemische Potential typischerweise:
für ideale Gase und ideale Mischungen. Das bedeutet, dass mit abnehmender Teilchendichte
eines Reaktanten das chemische Potential immer negativer wird,
was eine divergierende chemische Kraft erzeugt, die weiterer Reaktion entgegenwirkt.
Daher kann das System niemals einen Zustand erreichen, in dem
für eine beliebige Komponente. Stattdessen verlangsamt sich die Reaktion und stagniert asymptotisch, wenn sich der Verbrauch nähert.
Ähnlich können wir auch und definieren.
Das partielle molare Enthalpie und die partielle molare Entropie sind definiert als:
Diese beiden Variablen spielen die gleiche Rolle wie das chemische Potential bei der
Konstruktion der Gibbs-Energie.
und sind alle homogene Funktionen bezüglich ,
Durch Ableitung nach auf beiden Seiten erhalten wir:
Erinnern wir uns an , finden wir weiter:
Dann können wir, wie wir es für das chemische Potential und die Gibbs-Energie getan haben,
ähnliche Größen für und definieren:
und erhalten natürlich:
Nehmen wir an, die Reaktion ist thermodynamisch reversibel.
Betrachten Sie einen infinitesimalen Schritt einer chemischen Reaktion mit .
Die Reaktionswärme, d. h. die vom System während des Reaktionsschritts absorbierte Wärme, ist gegeben durch:
Wenn wir weiter annehmen, dass das System sehr nahe am Gleichgewicht ist,
verschwindet .
Dann reduziert sich die Wärme auf:
Das Gesetz der Massenwirkung liefert eine quantitative Beziehung zwischen
den chemischen Potentialen der reagierenden Spezies und ihren Konzentrationen (oder Aktivitäten) im chemischen Gleichgewicht. Es ist ein Ausdruck der Bedingung, dass die Gibbs-Energie minimiert wird, oder äquivalent, dass die chemischen Potentiale über die Reaktion ausgeglichen sind.
Das chemische Potential eines idealen Gases ist:
wobei die de-Broglie-thermische Wellenlänge ist.
Wählen wir als einen beliebigen Referenzdruck
und definieren als:
Dann:
wobei die Konzentration (Teilchendichte) ist.
Wir definieren die Aktivität :
Dann nimmt das chemische Potential die einheitliche Form an:
Nahe dem Gleichgewicht gilt:
Wir weisen diese Gleichung neu zu:
Definieren Sie weiter die Gleichgewichtskonstante:
Beachten Sie, dass die Gleichgewichtskonstante unabhängig von der Reaktionsdauer ist.
Diese Konstante ist auf dem Referenzzustand definiert.
Es gibt eine weitere ähnliche Größe,
die jedoch überall während einer Reaktion definiert ist,
die Reaktionsquotient genannt wird:
Sie sehen gleich aus, sind aber tatsächlich recht unterschiedlich.
Um dies zu verdeutlichen, verwenden wir :
Da im Gleichgewicht gilt,
sehen wir, dass diese Bedingung erfüllt.
2.9 Phasenübergang zweiter Ordnung
2.9.1 Ising-Modell
Ein Phasenübergang tritt auf, wenn sich die makroskopischen Eigenschaften eines Materials abrupt ändern, wenn ein externer Parameter, wie die Temperatur, variiert wird. Bei einem Phasenübergang erster Ordnung, exemplifiziert durch Phänomene wie das Sieden, bei dem sich die Dichte plötzlich ändert, weisen bestimmte thermodynamische Größen einen diskontinuierlichen Sprung am Übergangspunkt auf. Phasenübergänge zweiter Ordnung, auch kontinuierliche Phasenübergänge genannt, präsentieren ein anderes Bild. Hier ist eine fundamentale physikalische Größe, der Ordnungsparameter, zentral. Der Ordnungsparameter misst quantitativ den Grad der Ordnung oder der gebrochenen Symmetrie in der Struktur des Materials.
Insbesondere nimmt der Ordnungsparameter einen von Null verschiedenen Wert in der Phase an, die langreichweitige Ordnung aufweist, typischerweise bei niedrigeren Temperaturen, was die etablierte Ordnung signalisiert, wie die spontane Magnetisierung in einem Ferromagneten. Wenn sich die Temperatur bei einem Übergang zweiter Ordnung der kritischen Temperatur nähert, nimmt dieser Ordnungsparameter kontinuierlich und glatt ab und erreicht schließlich Null in der ungeordneten Hochtemperaturphase. Entscheidend ist, dass sich der Ordnungsparameter selbst kontinuierlich auf Null reduziert, während seine Ableitungen nach externen Parametern wie der Temperatur – Größen wie Suszeptibilität oder spezifische Wärme – typischerweise Diskontinuitäten aufweisen oder sogar am kritischen Punkt divergieren. Daher liegt das definierende Merkmal, das Phasenübergänge erster und zweiter Ordnung unterscheidet, im Verhalten des Ordnungsparameters: ein diskontinuierlicher Sprung signalisiert einen Übergang erster Ordnung, während eine kontinuierliche Abnahme auf Null einen Übergang zweiter Ordnung signalisiert.
In diesem Kapitel
verwenden wir Eisen als Beispiel für Phasenübergänge zweiter Ordnung.
Eisen zeigt unterhalb seiner kritischen Curie-Temperatur Ferromagnetismus,
wo spontane Magnetisierung auftritt,
während es oberhalb dieser Temperatur paramagnetisch wird.
Der fundamentale Ordnungsparameter, der diesen Übergang charakterisiert, ist die Magnetisierungsdichte,
bezeichnet als .
Betrachten Sie eine makroskopische Eisenprobe im thermischen Gleichgewicht mit einem Wärmebad bei Temperatur .
Um ein handhabbares statistisches Modell zu konstruieren,
teilen wir das System in kubische Gitterzellen auf.
Jede Zelle enthält ein thermodynamisch signifikantes Ensemble von Atomen –
ausreichend groß, dass statistische Mittelwertbildung gültig wird,
aber klein genug, um die räumliche Auflösung zu erhalten.
Innerhalb jeder Zelle
definieren wir die grobkörnige Magnetisierung als den Volumenmittelwert der atomaren Momente:
**Grobkörnigkeit**
Das Verfahren reduziert effektiv das komplexe Vielatomsystem auf eine Gitterdarstellung.
Entscheidend ist, dass in der ferromagnetischen Phase () zwei Schlüsseleffekte auftreten:
Lokale Ausrichtung: Austauschwechselwirkungen treiben eine nahezu perfekte parallele Ausrichtung der atomaren Momente innerhalb jeder Zelle an.
Spontane Symmetriebrechung: Die gesamte Zelle wählt spontan eine bevorzugte Magnetisierungsrichtung.
Diese kollektiven Phänomene rechtfertigen die Darstellung jeder grobkörnigen Einheit durch eine binäre Spinvariable ,
wobei:
- entspricht einer makroskopischen "Spin-up"-Orientierung.
- repräsentiert eine makroskopische "Spin-down"-Orientierung.
Die diskrete -Beschreibung erfasst die wesentliche Physik der gebrochenen Symmetrie und vernachlässigt untergeordnete Fluktuationen.
Diese Vereinfachung bewahrt universelles kritisches Verhalten nahe und ermöglicht leistungsfähige analytische und rechnerische Ansätze zur Modellierung von Phasenübergängen.
Nach der Grobkörnigkeit führen wir das **Ising-Modell** ein.
Sein Diagramm ist unten gezeigt:
Diagramm des Ising-Modells
Das Ising-Modell weist jedem Gitterplatz binäre Spins zu.
Spins wechselwirken mit ihren nächsten Nachbarn und dem externen Feld.
Dies ergibt die Hamilton-Funktion:
Hier zeigt ferromagnetische Kopplung und antiferromagnetische.
ist das externe Magnetfeld.
Da nur annehmen kann,
wenn mehr als 3 Spins betrachtet werden,
kollabiert die Gleichung schließlich zu einem Zweikörperterm,
der einer Zweikörperwechselwirkung entspricht.
Mittlerweile hat es eine Translationssymmetrie,
was bedeutet, dass die Hamilton-Funktion und die physikalischen Eigenschaften invariant bleiben
unter jeder diskreten Verschiebung aller Spins um einen Gitterplatz aufgrund der
gleichmäßigen Gitterstruktur und identischen Wechselwirkungen.
Unter Verwendung des kanonischen Ensembles
können wir die Zustandssumme schreiben:
wobei
Definieren Sie die Helmholtz-Energie:
Thermodynamische Observablen werden als Ableitungen von erhalten. Zum Beispiel ist die Magnetisierung pro Gitterplatz:
Überprüfen Sie gleichzeitig die Helmholtz-Freie Energie,
nehmen Sie die Ableitung nach dem externen Feld :
ist tatsächlich die Wahrscheinlichkeitsverteilung,
da wir uns im kanonischen Ensemble befinden.
Daher ergibt sich die obige Ableitung zu:
Dann kann die Magnetisierung durch die Ableitung von dargestellt werden:
Die magnetische Suszeptibilität,
die die Reaktion auf das externe Feld misst, ist:
Diese Gleichung zeigt, dass die magnetische Suszeptibilität
von der Fluktuation der Gesamtmagnetisierung abhängt.
Die Zweipunkt-Korrelationsfunktion misst, inwieweit Spins
an verschiedenen Orten korreliert sind:
In einem translationsinvarianten System
ist ,
genannt Korrelationsfunktion.
Dann ist die magnetische Suszeptibilität gegeben durch:
Sei eine Test-Wahrscheinlichkeitsverteilung über Spin-Konfigurationen
.
Erinnern wir uns, dass das Nichtgleichgewichts-Helmholtz-Freie Energie-Funktional definiert ist als:
Um zu minimieren, nehmen Sie die Variation,
Im Gleichgewicht gilt:
Wir können diese
Ungleichung verwenden, um eine Variationsnäherung für den Gleichgewichtszustand und
die Gleichgewichts-freie Energie zu finden.
Der nächste Schritt ist der entscheidende:
Wir ersetzen die tatsächlichen fluktuierenden Wechselwirkungen eines Spins durch ein effektives Feld, das durch die mittlere Magnetisierung seiner Nachbarn erzeugt wird.
Basierend auf dieser Annahme
werden die Wahrscheinlichkeiten jedes Spins entkoppelt und unabhängig.
Dann wird die gemeinsame Verteilung zum einfachen Produkt jeder Wahrscheinlichkeit:
Wir nehmen an, dass ,
da es Translationssymmetrie hat.
Für Letzteres sind Spins an benachbarten Stellen antiparallel zueinander, und
daher müssten wir zwei verschiedene Wahrscheinlichkeitsverteilungen für
zwei Subgitter einführen.
Für einen einzelnen Spin
haben wir zwei Nebenbedingungen:
Wenn wir dies lösen, erhalten wir:
Dann können wir mit einigen Ableitungen
die Bedingung finden, um zu minimieren:
wobei z die Koordinationszahl ist,
was bedeutet, wie viele Spins sich neben einem bestimmten Spin befinden.
Weil
Wir haben:
Und:
Dann:
Dies ist die Bedingung zur Minimierung von .
Leider können wir diese Gleichung nicht analytisch lösen.
Betrachten wir eine spezielle Situation mit .
Dann wird die Gleichung:
Wir können die Gleichung stattdessen untersuchen.
Wenn , hat 3 Lösungen;
und wenn , hat nur 1 Lösung.
Offensichtlich ist hier.
Wir können die kritische Situation sehen, wenn .
Daher können wir die kritische Temperatur leicht finden.
Wenn die Gleichung drei Lösungen ergibt,
entstehen zwei symmetrische Nicht-Null-Lösungen.
Wenn die Temperatur sinkt,
bestimmen anfängliche Fluktuationen, welchen Zweig das System wählt,
was zu spontaner Symmetriebrechung führt.
2.9.2 Skalierungshypothese
Bei Phasenübergängen zweiter Ordnung (kontinuierlich)
ändert sich die Symmetrie des Systems,
und kritische Phänomene treten nahe dem kritischen Punkt auf.
Diese Phänomene – einschließlich Potenzgesetz-Divergenzen,
Skalierungsgesetze, Universalität
und fraktales Verhalten – entstehen aus der Divergenz der Korrelationslänge und der Korrelationszeit.
Kritische Phänomene werden durch eine Reihe kritischer Exponenten charakterisiert,
die beschreiben, wie physikalische Größen als Potenzgesetze im reduzierten Temperatur-
(wobei die kritische Temperatur ist) oder im externen Feld verhalten.
Zur Analyse dieser Verhaltensweisen
werden physikalische Größen zerlegt in:
Ein singulärer (divergenter) Teil: Dieser Teil divergiert oder verschwindet als Potenzgesetz am kritischen Punkt.
Ein regulärer (glatter) Teil: Dieser Teil bleibt endlich und nicht-singulär.
Die kritischen Exponenten werden durch die Potenzgesetz-Skalierung der singulären Teile definiert. Unten sind die wichtigsten Potenzgesetz-Beziehungen, illustriert am Ising-Modell als Beispiel.
Spezifische Wärme ():
Der singuläre Teil der spezifischen Wärme divergiert als , wenn (d. h. wenn ). Für divergiert bei ; für kann sie eine logarithmische Divergenz aufweisen.
Ordnungsparameter ():
Unter () verschwindet die spontane Magnetisierung (Ordnungsparameter) als . Dies beschreibt, wie die geordnete Phase verschwindet, wenn von unten an herankommt.
Suszeptibilität ():
Die magnetische Suszeptibilität (Reaktion auf ein externes Feld) divergiert nahe als . Dies zeigt eine erhöhte Reaktion auf Störungen am kritischen Punkt an.
Kritische Isotherme ( bei ):
Bei () hängt die Magnetisierung vom externen Feld als Potenzgesetz ab. Ein großes impliziert eine schwache Reaktion auf am kritischen Punkt.
Korrelationsfunktion ():
Die Korrelationsfunktion beschreibt räumliche Korrelationen.
Die Korrelationslänge gibt an, wie weit ein Spin einen anderen Spin beeinflussen kann.
Am kritischen Punkt () zerfällt sie algebraisch als , wobei die räumliche Dimension ist. Der Exponent quantifiziert Abweichungen vom mittleren Feldzerfall.
Korrelationslänge ():
Die Korrelationslänge divergiert als nahe und definiert die räumliche Skala, über die Fluktuationen korreliert sind. Diese Divergenz ist die Grundlage aller kritischen Phänomene.
Die Exponenten sind universell –
sie hängen nur von der räumlichen Dimension und der Symmetrie (z. B. Ising-Klasse) ab,
nicht von mikroskopischen Details.
Physiker haben sie experimentell gemessen,
derzeit können Sie sie als experimentelle Schlussfolgerung betrachten.
Diese Potenzgesetze bilden einen grundlegenden Rahmen für das Verständnis kritischer Phänomene in verschiedenen Systemen,
von Magneten bis zu Flüssigkeiten.
Um zu erklären, warum die Potenzgesetze gelten,
stellt Landau die Skalierungshypothese auf.
Die Skalierungshypothese ist die zentrale Idee, die modernen Theorien der
kritischen Phänomene zugrunde liegt. Sie besteht aus zwei Hauptpostulaten:
Singulärer Teil der freien Energie. Nahe dem kritischen Punkt kann die freie
Energie in einen glatten Teil und einen singulären Teil zerlegt werden.
Der singuläre Teil wird als verallgemeinerte homogene Funktion von und angenommen:
wird als Skalierungsfunktion bezeichnet,
die glatt ist, außer bei .
Einzelne Längenskala: Nahe dem kritischen Punkt ist die Korrelationslänge, die einzige relevante Längenskala. Dies bedeutet, dass der Phasenübergang zweiter Ordnung darauf beruht, dass nahe dem kritischen Punkt die Korrelationslänge zu groß (divergent) wird, sodass alle mikroskopischen Skalen im Vergleich zur Korrelationslänge zu klein sind.
Daher wird jede Skalierung für das gesamte System die Eigenschaften des Systems nicht ändern.
Daher muss der singuläre Teil der freien Energie nur von der Temperatur
und der Korrelationslänge abhängen.
Nach Dimensionsanalyse
muss in -dimensionalem Raum die Einheit von haben.
Dann ist es natürlich zu finden:
Es stellt sich heraus, dass diese Annahme nur für gilt.
Betrachten wir zunächst die Implikation der Skalierungsform der singulären freien Energie:
Setzt man , so erfordert , um singuläres Verhalten zu gewährleisten, was zu Folgendem führt:
Die Wärmekapazität wird aus der zweiten Ableitung der freien Energie abgeleitet:
was als Exponenten der Wärmekapazität bestätigt.
Die Ableitung von nach ergibt die Magnetisierung:
Für und erfordert spontane Magnetisierung , was ergibt:
Eine weitere Ableitung ergibt die Suszeptibilität:
Bei gilt , was zu Folgendem führt:
Aus der Ein-Längen-Skala-Hypothese und :
Die Beziehung zwischen Suszeptibilität und Korrelation kombiniert mit für ergibt:
Diese ergeben fünf Skalierungsrelationen für die Exponenten
.
Durch Eliminierung von ergeben sich vier experimentell überprüfbare Relationen:
Fisher-Gesetz:
Rushbrooke-Gesetz:
Widom-Gesetz:
Josephson-Gesetz:
Daher sind nur zwei dieser Exponenten unabhängig.
2.9.3 Landau-Freie Energie
Kehren wir zur Ising-Hamilton-Funktion zurück:
und die kanonische Zustandssumme sowie die freie Energie:
Da ,
führen wir nun die Summe über die Spin-Konfigurationen in Gleichung (36) in zwei Schritten durch:
Zuerst summieren über alle Spin-Konfigurationen mit festem .
Dann summieren über alle zulässigen Werte von .
Die Zustandssumme kann dann geschrieben werden als:
wobei die Zustandssumme eines Teil-Ensembles ist, bei dem fest ist.
Dieses Ensemble hält die Gesamtmagnetisierung fest und summiert über Mikrozustände, die mit dieser Einschränkung kompatibel sind.
Die Landau-Freie Energie kann dann definiert werden:
Und die freie Energie im gesamten -Ensemble ist gegeben durch:
Im thermodynamischen Limes sind sowohl als auch extensive Größen.
Daher wird die Summe über M von nur einem Term dominiert: dem Wert von
, der den Exponenten maximiert.
Da dieser Term allen anderen Termen weit überlegen ist, selbst ihrer Summe.
Diese Operation wird als
Sattelpunkt-Approximation bezeichnet.
Dann:
Um den Wert von in dieser Situation zu finden, bezeichnet als :
Diese Gleichung gibt tatsächlich die Zustandsgleichung an.
2.9.4 Landau-Theorie der Phasenübergänge
Wir definieren die freie Energie pro Spin und die Landau-Freie Energie pro Spin:
Gemäß Abschnitt 2.9.3:
Es ist Landau's Idee, als Taylor-Reihe von zu entwickeln:
Ungerade Terme verschwinden, da wir verlangen, dass die Energie unverändert bleibt,
wenn sich in seiner Richtung ändert (Symmetrie).
In der obigen Gleichung
wird durch den Hintergrund verursacht,
ändert sein Vorzeichen nahe .
Nahe dem kritischen Punkt,
können wir die reduzierte Temperatur
und einführen, um zu vereinfachen.
Wenn (), , und wir können den quartischen Term in der Landau-Freien Energie ignorieren, da .
Das Minimum tritt bei auf,
und das Zurücksubstituieren ergibt:
Offensichtlich sind und jeweils der reguläre Teil und
der singuläre Teil der freien Energie.
Wir müssen die singuläre freie Energie in die Skalierungsform bringen.
Die obige Gleichung ergibt .
Um die Koeffizienten auszugleichen,
Dies ergibt
Jedoch definiert das Verhalten von .
Aus der exakten Form von ,
wenn , verschwindet der singuläre Teil.
Dann können wir nur eine Lösung haben:
Beachten Sie, dass unter solchen Parameterkonfigurationen konvergiert.
Dies ist tatsächlich die Grenze der Landau-Theorie.
Wenn (), aus der obigen Analyse ist immer der reguläre Teil der freien Energie. Wir können ihn daher in der Legendre-Transformation ignorieren und uns direkt auf die singuläre freie Energie konzentrieren. Für wird die singuläre freie Energie durch Minimierung abgeleitet:
Um die Skalierungsform zu extrahieren, skalieren wir die Variablen neu. Definieren Sie die dimensionslose Magnetisierung und das Feld:
Das Einsetzen in den Ausdruck für die freie Energie ergibt:
wobei eine dimensionslose Funktion ist, die durch das dimensionslose Minimierungsproblem bestimmt wird. Dies stellt die Skalierungsform wieder her:
was die kritischen Exponenten bestätigt:
Andere Exponenten können ebenfalls durch die Landau-Funktion berechnet werden.
Exponent der spontanen Magnetisierung :
Bei und ergibt die Minimierung der Landau-Freien Energie:
Somit ist .
Exponent der kritischen Isotherme :
Bei ergibt die Minimierung:
Daher ist .
Exponent der Suszeptibilität :
Für () ergibt die lineare Antwort:
Somit ist .
Exponenten der Korrelationslänge und der Korrelationsfunktion :
Unter Verwendung der Gaußschen Approximation des Landau-Ginzburg-Funktionals:
Die Korrelationsfunktion erfüllt:
mit der Lösung:
Dies impliziert . Das Kurzstreckenverhalten entspricht (da impliziert ).
Verifizierung durch Fisher-Gesetz: Die Konsistenz wird bestätigt durch:
Zusammenfassung der Mittelwert-Feld-Exponenten:
Alle kritischen Exponenten in der Landau-Theorie sind:
Diese erfüllen die Skalierungsrelationen und sind exakt für räumliche Dimensionen .
Teil III. Quantenstatistik
3.1 Fermionen und Bosonen
3.1.1 Fundamentale Eigenschaften
Fermionen – zu denen Elektronen, Protonen, Neutronen und Quarks gehören – sind Quantenteilchen mit halbzahligen Spin, deren Wellenfunktionen unter Teilchenaustausch antisymmetrisch sind. Diese definierende Eigenschaft ergibt sich aus dem Spin-Statistik-Theorem und prägt ihr Verhalten grundlegend:
Fermionische Antisymmetrie: Die Wellenfunktion kehrt ihr Vorzeichen um, wenn zwei beliebige Teilchen vertauscht werden:
Pauli-Ausschlussprinzip: Fermionen weigern sich, identische Quantenzustände zu besetzen, was zu räumlicher Ausschließung und Entartungsdruck in Neutronensternen und Weißen Zwergen führt.
Bosonen – einschließlich Photonen, Gluonen und Higgs-Bosonen – besitzen ganzzahligen Spin und weisen unter Teilchenaustausch symmetrische Wellenfunktionen auf:
Bosonische Symmetrie: Die Wellenfunktion bleibt unter Teilchenaustausch invariant:
Bose-Anreicherung: Mehrere Bosonen sammeln sich in Zuständen niedriger Energie an, was Phänomene wie Supraleitung und Bose-Einstein-Kondensation ermöglicht.
Quantenteilchen gehorchen auch dem Prinzip der Ununterscheidbarkeit, was die Quantenstatistik fundamental von der klassischen Mechanik unterscheidet. Während klassische Teilchen ihre Identität durch Trajektorien beibehalten, verlieren Quantenteilchen ihre Individualität – ihre Wellenfunktionen verschmelzen zu kollektiven Zuständen. Der Permutationsoperator , der die Teilchen und vertauscht, verkörpert dieses Prinzip:
Entscheidend in 3+1 Dimensionen erfüllt :
was erfordert, dass physikalische Zustände sich transformieren als:
Diese Phasenbeschränkung kristallisiert das Spin-Statistik-Theorem:
Wenn es zwei Teilchen in einem System gibt, sind Fermionen durch das Pauli-Ausschlussprinzip eingeschränkt, während Bosonen dies nicht sind:
Fermionen:
Dieser Zustand verschwindet, wenn , was das Pauli-Ausschlussprinzip manifestiert, das verhindert, dass Fermionen Quantenzustände teilen.
Bosonen:
Wenn , verstärkt sich dieser Zustand selbst und verdoppelt die Amplitude für Bosonen, sich in identischen Konfigurationen zu sammeln.
Und nun erweitern wir das System auf Teilchen. Der gesamte Zustandsraum wird zu Produkten von Hilbert-Räumen, genannt Fock-Raum:
In diesem Fock-Raum kann der gemeinsame Zustand aller Teilchen als Determinante oder Permanenz dargestellt werden.
Für Fermionen:
Die Antisymmetrie der Determinante gewährleistet die Vorzeichenumkehr der Wellenfunktion bei Teilchenaustausch und erzwingt automatisch den Ausschluss, wenn Zustände zusammenfallen.
Für Bosonen:
wobei bedeutet, alle negativen Vorzeichen durch positive zu ersetzen.
Die Summe über alle Permutationen konstruiert eine Wellenfunktion, die unter Teilchenaustausch invariant ist und die unbegrenzte Besetzung von Einteilchenzuständen ermöglicht.
Diese Symmetriedichotomie orchestriert Quanten-Vielteilchenphänomene – von der Stabilität der Materie, die durch fermionische Ausschließung erzwungen wird, bis hin zu den kohärenten Quantenphasen, die durch bosonische Kondensation ermöglicht werden.
3.1.2 Zweite Quantisierung
Angenommen, wir haben sechs Teilchen, die auf Einteilchenzustände verteilt sind, die durch Impuls- oder Energie-Eigenzustände gekennzeichnet sind:
Stellen Sie sich vor:
Teilchen 1 im Zustand
Teilchen 2,3 im Zustand
Teilchen 4,5,6 im Zustand
Der entsprechende Produktzustand ist:
und wir müssen dann den Produktzustand symmetrisieren (für Bosonen) oder antisymmetrisieren (für Fermionen), was mühsam und umständlich ist. Am Ende dieses Prozesses interessieren wir uns jedoch nur für die Anzahl der Teilchen in jedem Einteilchenzustand. In diesem Fall kombinieren wir die gleichen Zustände und bezeichnen nur die Besetzungszahlen:
wobei die Anzahl der Teilchen im Zustand darstellt. Zum Beispiel wird die obige Konfiguration als
bezeichnet.
Für Bosonen kann beliebige nicht-negative ganze Zahlen annehmen. Fermionen dürfen jedoch Pauli nicht verletzen. Daher können von Fermionen nur 0 oder 1 annehmen.
3.1.3 Erzeugungs- und Vernichtungsoperatoren
Wir sind nun bereit, die natürlichen Operatoren einzuführen, die auf diese Zustände wirken: Erzeugungs- und Vernichtungsoperatoren. Diese Operatoren tun genau das, was ihre Namen andeuten:
: erzeugt ein Teilchen im Einteilchenzustand : vernichtet ein Teilchen aus dem Zustand
Ihre Wirkung auf die Besetzungszahlbasis ist wie folgt definiert:
Diese Definitionen gewährleisten die korrekte Normierung der Zustände und erhalten die Orthonormalität der Fock-Basis.
Die Algebra dieser Operatoren hängt von der Statistik der Teilchen ab. Traditionell verwenden wir für Bosonen und für Fermionen. Sie haben die folgenden Eigenschaften:
Für Bosonen:
Für Fermionen:
Hier bezeichnet den Kommutator und den Antikommutator. Wir definieren weiter die Operator-Zahlen:
für Bosonen
für Fermionen
Um dies zu überprüfen, wenden Sie diesen Operator einfach auf einen Zustand an, und Sie erhalten schließlich einen Eigenwert von .
Wir können leicht die folgenden Kommutationsrelationen zeigen:
Für Bosonen:
Für Fermionen:
Diese Operatoren bieten eine mächtige und elegante Sprache für Quanten-Vielteilchensysteme. Sie:
Vereinfachen die Konstruktion von Vielteilchenzuständen und Operatoren
Kodieren die Teilchenstatistik algebraisch
Bilden die Grundlage für Quantenfeldtheorie und Quanten-Vielteilchentheorie
Betrachten Sie zum Beispiel den Operator:
der in der Sprache der zweiten Quantisierung völlig natürlich ist. Es ist ein linearer Operator auf dem Fock-Raum, und wenn er auf das Vakuum angewendet wird, ergibt er einen gültigen physikalischen Zustand:
Wir können Erzeugungsoperatoren verwenden, um die Besetzungszahl-Basiszustände aus dem Vakuumzustand zu konstruieren:
3.2 Freies Fermi-Gas
3.2.1 Thermodynamische Eigenschaften
Für ein makroskopisches freies Fermi-Gas erfordert die riesige Teilchenzahl die Verwendung der zweiten Quantisierung, um den Zustand des Systems darzustellen; dieser Formalismus beinhaltet inhärent die Welle-Teilchen-Dualität von de Broglie, bei der Teilchen sowohl Wellencharakter (ebene Wellen) als auch Granularität aufweisen. Um die Gesamtenergie dieses nicht wechselwirkenden Gases zu bestimmen, verwenden wir den Hamilton-Operator der zweiten Quantisierung:
Beachten Sie, dass jedes mehr als 1 Zustand hat, da wir die Spins berücksichtigen müssen. Im Allgemeinen enthält jede Energie mit Spin Zustände. Die wird als Entartung bezeichnet.
Bevor wir fortfahren, ist es wichtig zu verstehen, wann Quanteneffekte für ein Gas wichtig werden. Bei hohen Temperaturen oder niedrigen Dichten verhält sich das Gas klassisch, und die Quantenstatistik kann ignoriert werden. Wenn jedoch die thermische de-Broglie-Wellenlänge
vergleichbar mit dem mittleren interpartikulären Abstand wird, überlappen sich die thermischen Wellenpakete einzelner Teilchen erheblich, und daher können Quanteneffekte nicht mehr vernachlässigt werden. Der mittlere interpartikuläre Abstand ist ungefähr , wobei die Teilchenzahldichte ist. Daher tritt Quantenentartung ein, wenn
Dies ergibt schließlich die kritische Temperatur, bei der Quanteneffekte nicht einfach vernachlässigt werden können. Für Fermionen wird sie als Fermi-Temperatur bezeichnet. Oder, in diesem Teil, wird auf 1 normiert, sodass Temperatur und Energie die gleiche Einheit haben. Dieser Wert kann auch die Fermi-Energie sein:
Wenn , spielen Quanteneffekte eine wichtige Rolle, und umgekehrt.
Wir setzen unsere Untersuchung des Fermi-Gases mit dem großkanonischen Ensemble fort, da die Teilchenzahl nicht konstant bleibt. In einem Quantensystem wird die großkanonische Zustandssumme durch die Spur berechnet:
wobei . Die Spur berücksichtigt alle möglichen Eigenwerte von und . Und für Fermionen gilt . Daher:
Also:
Durch Einsetzen von erhält man die Wahrscheinlichkeit, dass die Werte 0 und 1 annimmt. Wir können nun die durchschnittliche Besetzungszahl eines gegebenen Einteilchenzustands berechnen.
Dies wird als Fermi-Dirac-Verteilung bezeichnet.
Darüber hinaus können wir das großkanonische Potential erhalten:
Summieren über die Spins zur Vereinfachung:
Mithilfe des großkanonischen Potentials können wir alle thermodynamischen Größen ableiten:
Die durchschnittliche Teilchenzahl:
Die innere Energie:
Der Druck:
3.2.2 Zustandsdichte (DOS)
Im thermodynamischen Limes, wo das Volumen sehr groß wird, bilden die erlaubten Impulse eine dichte Menge. Es ist dann praktisch, die Summe über diskrete durch ein Integral über den kontinuierlichen Impulsraum zu ersetzen:
wobei das Volumen pro diskretem -Punkt im -Raum ist (da der Abstand zwischen benachbarten -Punkten in jeder Dimension unter periodischen Randbedingungen beträgt, was das Volumen pro Zustand im -Raum zu macht. Der Kehrwert davon ergibt den Vorfaktor).
Bei der Betrachtung von energieabhängigen physikalischen Größen (z. B. Berechnung der Zustandssumme, Gesamtenergie oder Wärmekapazität in der statistischen Mechanik) ist es weiterhin nützlich, das Konzept der Zustandsdichte (DOS) einzuführen. Die Zustandsdichte ist definiert als die Anzahl der Zustände pro Energieintervall, was bedeutet, dass die Anzahl der Zustände mit Energie zwischen und darstellt. Dies ermöglicht es uns, das Impulsintegral in ein Energieintegral umzuwandeln, was die Berechnungen vereinfacht.
Um abzuleiten, nutzen wir die Eigenschaften isotroper Systeme (wie das freie Elektronengas), bei denen die Energie nur von der Größe des Wellenvektors abhängt, über die Beziehung . Ausgehend vom Impulsraum-Integral wird die differentielle Anzahl der Zustände geschrieben als:
In Kugelkoordinaten gilt . Die Integration über den Winkelteil ergibt:
somit:
Variablenumwandlung unter Verwendung der Energie-Impuls-Beziehung:
also:
Einsetzen in :
Die Zustandsdichte ist definiert als , daher:
Dies wird oft in der vereinfachten Form geschrieben:
Mithilfe der Zustandsdichte kann jede Summe über (wenn der summierte Term nur von der Energie abhängt) in ein Integral über die Energie umgewandelt werden:
Dies ist äußerst effizient bei der Behandlung thermodynamischer Größen (wie der Fermi-Dirac-Verteilung für ein Elektronengas), da es die Dimensionalität des Integrals reduziert und die Energieabhängigkeit hervorhebt.
Mit der Zustandsdichte können wir die thermodynamischen Größen neu formulieren:
Großkanonisches Potential
Teilchenzahl
Innere Energie
Wir können durch Teile integrieren und schließlich zeigen:
was eine exakte Beziehung zwischen dem Druck und der inneren Energiedichte liefert:
3.2.3 Null-Temperatur-Grenze
Wenn sich die Temperatur 0 nähert, reduziert sich die Fermi-Dirac-Verteilungsfunktion zu einer Einheitsstufenfunktion um die Energie .
Und das chemische Potential bei wird als Fermi-Energie definiert:
Daher sind bei alle Einteilchenzustände mit Energie kleiner als besetzt und alle Zustände mit Energie größer als leer. Diese Konfiguration bildet eine Fermi-Kugel im -Raum, deren Radius ist, wodurch die Zustände auf der Oberfläche zu werden.
Fermi-Kugel,
Dann ist die Teilchenzahl gegeben durch:
Einsetzen der Zustandsdichte aus dem letzten Abschnitt:
Lösen nach ergibt:
Für Elektronen gilt , daher stimmt die aus der Null-Temperatur-Grenze berechnete mit der aus der de-Broglie-Wellenlänge berechneten überein.
Wir können die Zustandsdichte mithilfe der Fermi-Energie neu schreiben:
Die innere Energie wird:
Kombinieren der obigen Gleichungen ergibt:
was besagt, dass bei Null Temperatur die durchschnittliche Energie pro Teilchen proportional zur Fermi-Energie ist.
Schließlich kann der Druck erhalten werden:
Daher übt das Fermi-Gas auch bei Null Temperatur einen von Null verschiedenen Druck aus, bekannt als Entartungsdruck. Dieser Druck ist auf das Pauli-Ausschlussprinzip zurückzuführen. Dieser Entartungsdruck spielt eine entscheidende Rolle für die Stabilität dichter astrophysikalischer Objekte wie Weißer Zwerge und Neutronensterne. Diese Sterne kollabieren, wenn ihre Entartungsdrücke nicht stark genug sind, um dem negativen Druck der Gravitationskraft standzuhalten.
3.2.4 Niedrigtemperatur-Sommerfeld-Expansion
Bei niedrigen, aber endlichen Temperaturen ist die Fermi-Dirac-Verteilung keine scharfe Stufenfunktion mehr. Stattdessen wird sie um die Fermi-Energie leicht verschmiert. Wir werden diese kleine Abweichung mit der Sommerfeld-Expansion untersuchen.
Angenommen, wir möchten Integrale der Form berechnen:
Die Schlüsselidee, die die Sommerfeld-Expansion ermöglicht, ist die Erkenntnis, dass die Ableitung der Fermi-Dirac-Verteilung, , als thermisch verbreiterte Dirac-Delta-Funktion fungiert, die bei zentriert ist und eine Breite von hat. Diese Funktion, definiert als:
ist auf Eins normiert und wird bei scharf bei konzentriert. Um dieses Verhalten auszunutzen, integrieren wir partiell nach Einführung der Hilfsfunktion . Dies ergibt:
Die Randterme verschwinden: Bei ist ; bei geht exponentiell. Somit:
Im Niedrigtemperatur-Limes () kann die untere Integrationsgrenze auf erweitert werden, da für schnell abfällt. Entwicklung von in eine Taylor-Reihe um :
und Einsetzen in das Integral ergibt:
Die Integrale (mit ) verschwinden für ungerade aufgrund der Antisymmetrie. Für gerade ergeben sie sich zu:
wobei die dimensionslosen Integrale mit der Riemannschen Zeta-Funktion zusammenhängen:
Unter Verwendung von und sind die ersten Koeffizienten:
Da für , wird die Reihe:
Diese Expansion ist asymptotisch, wobei jeder Term proportional zu ist. Die Dominanz der Fermi-Oberfläche () bei niedrigen Temperaturen gewährleistet eine schnelle Konvergenz, wenn .
Aufbauend auf der Sommerfeld-Expansionstechnik untersuchen wir nun systematisch, wie sich wichtige thermodynamische Größen des entarteten Fermi-Gases bei niedrigen Temperaturen entwickeln. Die Expansion bietet einen leistungsfähigen Rahmen zur Berechnung präziser temperaturabhängiger Korrekturen zum Verhalten bei Null Temperatur.
Chemisches Potential
Die grundlegende Verbindung zwischen Teilchenzahl und chemischem Potential ergibt sich aus der Zustandsdichte und der Fermi-Dirac-Verteilung:
was sich zur Einschränkungsgleichung vereinfacht:
Anwendung der Sommerfeld-Expansion mit ergibt:
Die perturbative Lösung durch Schreiben von und Entwicklung in kleine und ergibt die Temperaturabhängigkeit des chemischen Potentials:
Dies zeigt, dass quadratisch von mit steigender Temperatur abnimmt, was die thermische Anregung von Fermionen knapp unterhalb der Fermi-Oberfläche widerspiegelt.
Innere Energie
Der Ausdruck für die innere Energie:
wird unter Verwendung von in der Sommerfeld-Expansion ausgewertet:
Einsetzen des chemischen Potentials von oben und erneutes Ausdehnen in ergibt:
Der quadratische Anstieg von mit der Temperatur steht im scharfen Gegensatz zu klassischen Gasen und entsteht durch die Pauli-Blockade, die Anregungen auf eine dünne Schale nahe beschränkt.
Druck und Kompressibilität
Unter Verwendung der Beziehung aus dem großkanonischen Potential erbt der Druck die Temperaturabhängigkeit der inneren Energie:
Die isotherme Kompressibilität folgt dann aus ihrer thermodynamischen Definition:
Durch Differenzieren von unter Berücksichtigung von ergibt sich:
Dies zeigt, dass die Kompressibilität mit der Temperatur abnimmt, was darauf hindeutet, dass das Gas steifer wird, wenn thermische Anregungen Zustände oberhalb der Fermi-Oberfläche besetzen.
3.3 Freies Bose-Gas
3.3.1 Thermodynamische Eigenschaften
In dieser Vorlesung untersuchen wir ein System von nicht wechselwirkenden massiven bosonischen Teilchen, die in einem Volumen bei Temperatur und chemischem Potential eingeschlossen sind. Bosonen gehorchen der Bose-Einstein-Statistik, was bedeutet, dass mehrere Teilchen denselben Quantenzustand besetzen können. Dies steht im Gegensatz zu Fermionen, die dem Pauli-Ausschlussprinzip gehorchen, das verhindert, dass mehr als ein Teilchen denselben Quantenzustand besetzt.
Unser Forschungsweg ist derselbe wie bei Fermionen.
Im großkanonischen Ensemble gilt weiterhin:
Dann sind alle thermodynamischen Größen gegeben:
Großkanonisches Potential
Es ist lehrreich, als Produkt unendlich vieler großkanonischer Zustandssummen für Einteilchenzustandssysteme zu schreiben:
Ähnlich kann das großkanonische Potential als Summe geschrieben werden:
Diese Ergebnisse zeigen, dass das Bose-Gas als eine unabhängige Summe unendlich vieler kleiner offener Systeme verstanden werden kann. Diese kleinen offenen Systeme stehen im Kontakt mit demselben Bad mit Temperatur und chemischem Potential .
Teilchenzahl
Dies ist als Bose-Einstein-Verteilung bekannt.
Wir haben auch:
Währenddessen bleibt die Zustandsdichte für Fermionen für Bosonen korrekt, da sie auf die gleiche Weise abgeleitet wird.
3.3.2 Hochtemperatur-Grenze
Bei hohen Temperaturen oder niedrigen Dichten verhalten sich Quantengase klassisch, wobei Quantenstatistik-Effekte als kleine Korrekturen erscheinen. Der Schlüsselparameter, der diese Korrekturen steuert, ist der Entartungsparameter
wobei die Teilchendichte und
die thermische de-Broglie-Wellenlänge ist. Wenn , kann die Quantenstatistik perturbativ behandelt werden. Für eine einheitliche Beschreibung von Bose- und Fermi-Gasen führen wir den statistischen Parameter ein:
Dies ermöglicht es uns, die Besetzungszahlverteilung und das großkanonische Potential in einer einheitlichen Form auszudrücken. Die Bose-Einstein-Funktionen () und Fermi-Dirac-Funktionen () werden durch Integralrepräsentationen definiert, die Potenzreihenentwicklungen in der Fugazität zulassen:
die für konvergieren.
Mit der Zustandsdichte können mit einem Integral geschätzt werden:
wobei .
Die Teilchenzahldichte und Energiedichte nehmen dann kompakte Formen an:
wobei die Spin-Entartung ist. Die Zustandsgleichung ergibt sich als für beide Statistiken. Um Hochtemperatur-Entwicklungen abzuleiten, lösen wir die Teilchenzahlgleichung perturbativ nach kleinem . Das Invertieren der Reihe
ergibt die Fugazitätsentwicklung:
Durch Einsetzen dieser Werte in die Druck- und Energieausdrücke und erneutes Ausdehnen in Potenzen von erhält man die Quantenkorrekturen zum klassischen Verhalten:
Die Entwicklung des chemischen Potentials folgt ähnlich:
Der Vorzeichenunterschied in den -Termen offenbart grundlegendes statistisches Verhalten: die bosonische Austausch-Symmetrie reduziert Druck und Energie im Vergleich zu klassischen Vorhersagen, während die fermionische Ausschließung sie erhöht. Dies spiegelt wider, wie Quantenstatistiken effektive Wechselwirkungen zwischen Teilchen modifizieren – Bosonen zeigen eine effektive Anziehung, während Fermionen eine effektive Abstoßung im Vergleich zu unterscheidbaren Teilchen aufweisen. Die Genauigkeit dieser Entwicklungen wird durch die Kleinheit von gesteuert, die die Überlappung thermischer Wellenpakete misst und im klassischen Limes verschwindet.
3.3.3 Bose-Einstein-Kondensation
Kehren wir zur Bose-Einstein-Verteilung zurück.
Damit die durchschnittliche Besetzungszahl positiv ist, muss das chemische Potential kleiner als das Energieniveau sein. Dies muss für jeden Einteilchenzustand gelten. Daher haben wir:
wobei der Einteilchen-Grundzustand ist. Für ein freies Bose-Gas, das in einer Box eingeschlossen ist, haben wir:
Daher haben wir:
Wenn , divergiert die Teilchenzahl im Grundzustand offensichtlich. Für ein System mit endlicher Teilchenzahl und endlichem Volumen kann eine solche Divergenz natürlich nicht auftreten. Wir müssen andere physikalische Bedingungen heranziehen, um die tatsächliche Anzahl der Teilchen im Grundzustand zu bestimmen. Wie wir als nächstes zeigen werden, kann in drei Dimensionen ein makroskopischer Bruchteil der Teilchen den Grundzustand besetzen, ein Phänomen, das als Bose-Einstein-Kondensation (BEC) bezeichnet wird.
Zur Vereinfachung betrachten wir spinlose Bosonen, also . Unter Verwendung der Entwicklung erhalten wir:
Da , ist die Fugazität . Wenn von 0 auf 1 ansteigt, nähert sich die Bose-Einstein-Funktion einem endlichen Wert, dargestellt durch die Riemannsche Zeta-Funktion:
Hier gibt es einen Konflikt. Betrachten wir eine feste Temperatur und erhöhen allmählich die Anzahl der Teilchen im System. Die Teilchendichte erreicht allmählich den Sättigungspunkt:
Jedoch hat die linke Seite keine obere Grenze, während die rechte Seite eine hat.
Wir können auch die Teilchenzahl fixieren und finden die gleiche kritische Temperatur . Für hat die Gleichung keine Lösung.
Derzeit haben wir keine Ahnung, warum die Teilchenzahl eine Obergrenze hat. Diese Gleichung führt zu einer kritischen Zahl . Vielleicht brauchen wir einen Glaubenssprung.
Der Sprung wurde von Einstein vollbracht. Bei kritischen Punkt haben wir , also . Aber die Bose-Verteilung sagt uns, dass die Besetzungszahl des Grundzustands () ist:
Diese Divergenz ist ein Alarm, dass wir die diskrete Summe nicht wirklich als kontinuierliches Integral annähern können. Die Teilchenzahl im Grundzustand muss separat behandelt werden. Hier kommt Einsteins Genie zum Einsatz!
Einstein erklärte schließlich den integral-approximierten Teil der Summation neu:
Wenn wir das Integral der Zustandsdichte zur Schätzung der Summe verwenden.
Die Zustandsdichte verschwindet für . Währenddessen hat der Grundzustand Null Energie. Daher berücksichtigt das Integral den Grundzustand nicht. Dies ist, wie unser Glaube springt:
Wir betrachten es nur als die Teilchen der angeregten Zustände anstelle aller Bosonen, und Bosonen haben endliche angeregte Zustände. Wenn die Teilchen die Obergrenze der Anzahl der angeregten Zustände überschreiten, unabhängig von der Energie, muss der überschüssige Teil im Grundzustand aufgefüllt werden. Sie kondensieren im Grundzustand und tragen nichts zu den Eigenschaften des gesamten Systems bei. Dieses Phänomen wird Bose-Einstein-Kondensation genannt.
Unter dieser Perspektive können wir die obigen Gleichungen neu interpretieren:
Dies ist die maximale Kapazität aller angeregten Zustände. Unter dieser Zahl neigen Teilchen dazu, die angeregten Zustände zu füllen. Über diese Zahl hinaus müssen andere Teilchen in den Grundzustand übergehen.
Die Gesamtzahl:
Nach dieser Interpretation wird die Besetzungszahl des Grundzustands makroskopisch – proportional zum Systemvolumen.
Wir können die Teilchendichte weiter mit der Temperatur interpretieren, da die de-Broglie-Wellenlänge mit zusammenhängt. Wenn , tritt BEC auf, dann:
Entsprechend:
Wenn , keine BEC, , und die beiden Gleichungen gelten nicht mehr.
Aufgrund der Kondensation teilt sich das System in zwei Phasen auf:
Kondensat: Eine makroskopische Anzahl von Teilchen besetzt den Grundzustand . Diese Teilchen haben keine kinetische Energie und keinen Impuls. Infolgedessen tragen sie nichts zur Energie, zum Druck oder zur Entropie des Systems bei.
Thermische Wolke: Alle anderen Teilchen sind auf angeregte Zustände mit verteilt, beschrieben durch die übliche Bose-Einstein-Verteilung mit . Diese Teilchen bestimmen alle thermodynamischen Eigenschaften: Energie, Druck und Wärmekapazität.
Wenn , existiert das Bose-Gas vollständig in der thermischen Phase ohne Kondensat (). Die Fugazität , und alle thermodynamischen Eigenschaften werden von der thermischen Wolke bestimmt – Teilchen, die über angeregte Zustände () gemäß der Bose-Einstein-Verteilung verteilt sind. Für spinlose Bosonen () sind die Schlüsselbeziehungen:
Hier bezeichnen , und die Teilchendichte, die innere Energiedichte und den Druck der thermischen Wolke. Um die Zustandsgleichung zu bestimmen, löst man die erste Gleichung nach auf und setzt sie in die Ausdrücke für und ein.
Isotherme Kompressibilität
Die thermische Wolke zeigt kritisches Verhalten nahe . Die isotherme Kompressibilität quantifiziert ihre Reaktion auf Druckänderungen bei konstanter Temperatur:
Bei festem ergeben sich Änderungen in und allein aus Änderungen in über :
Unter Verwendung der Identität werden diese zu:
Im logarithmischen Format ():
Das Verhältnis ergibt :
wobei durch bestimmt ist. Wenn , geht und divergiert, während endlich bleibt. Folglich divergiert :
Diese Divergenz bedeutet kritische Fluktuationen – ein Kennzeichen kontinuierlicher Phasenübergänge. Die thermische Wolke wird nahe unendlich komprimierbar, was langreichweitige Korrelationen und eine erhöhte Empfindlichkeit gegenüber externem Druck widerspiegelt, obwohl andere thermodynamische Eigenschaften analytisch bleiben.
3.4 Eingefangen in einem Potential
Das Verhalten von Quantengasen, die in externen Potentialen eingeschlossen sind, zeigt grundlegende Unterschiede zu Systemen im freien Raum, wobei harmonische Fallen eine Eckpfeiler für Kaltatomexperimente darstellen. Für Bosonen in solchen isotropen Fallen erzeugt das diskrete Einteilchenspektrum
eine Zustandsdichte, die skaliert wie , was von der freien Raum-Abhängigkeit abweicht. Diese Umstrukturierung verändert die Bose-Einstein-Kondensation (BEC) dramatisch: die kritische Temperatur skaliert als
schwächer als die -Skalierung in homogenen Systemen. Unterhalb von folgt der Kondensatanteil
der schneller abfällt als das freie Raum-Gesetz aufgrund der spektralen Geometrie der Falle. Das Kondensat – lokalisiert im Grundzustand – trägt keine Energie und keinen Impuls bei, während die Thermodynamik vom thermischen Wolke gesteuert wird, die eine divergierende Kompressibilität nahe dem Übergang aufweist.
Für Fermionen in identischen Fallen tritt Entartung auf, wenn . Die Fermi-Energie bei Null Temperatur,
und die Grundzustandsenergie spiegeln das Gleichgewicht des Virialsatzes zwischen kinetischer und potentieller Energie wider. Bei niedrigen Temperaturen ergibt die Sommerfeld-Expansion
was freie Fermionen widerspiegelt, aber mit skalierten Exponenten. Die Einschränkung verteilt den Phasenraum neu und verändert Skalierungsrelationen – der Entartungsdruck folgt nun gegenüber im freien Raum.
Im semiklassischen Regime konvergieren diskrete Summen zu Phasenraum-Integralen:
Dies offenbart universelle Skalierungsgesetze wie
für -dimensionale Fallen, was zeigt, wie die Einschränkung die Thermodynamik umgestaltet und gleichzeitig Quantenentartungsphänomene beibehält. Kaltatomexperimente validieren diese Vorhersagen direkt und untersuchen das Zusammenspiel zwischen Quantenstatistik und räumlichen Einschränkungen.
3.5 Masseloses Quantengas
3.5.1 Zustandsdichte
Masselose bosonische Teilchen wie Photonen und Phononen weisen eine lineare Dispersionsrelation auf, wobei die Ausbreitungsgeschwindigkeit ist (Lichtgeschwindigkeit für Photonen, Schallgeschwindigkeit für Phononen). Um die Energiezustandsdichte im -dimensionalen Raum abzuleiten, betrachten wir die Anzahl der Quantenzustände in einer Kugelschale des Radius und der Dicke im -Raum. Das Volumenelement ist proportional zur Oberfläche einer -dimensionalen Kugel:
die Oberfläche einer Einheitskugel ist. Einsetzen der linearen Dispersion und ergibt:
Für Photonen in drei Dimensionen (), einschließlich zweier Polarisationszustände, gilt . Für Phononen in 3D-Kristallen, unter Berücksichtigung von drei akustischen Zweigen (zwei transversale, einer longitudinal), gilt .
3.5.2 Schwarzkörperstrahlung
Das elektromagnetische Feld im thermischen Gleichgewicht wird als ein Gas von nicht wechselwirkenden Photonen mit Null chemischem Potential modelliert. Jeder Modus mit Frequenz verhält sich wie ein quantenmechanischer harmonischer Oszillator mit durchschnittlicher Energie . Unter Verwendung der Photonenzustandsdichte ist die spektrale Energiedichte pro Volumeneinheit:
Die Integration über alle Frequenzen ergibt die **Gesamtenergiedichte** , die als skaliert (Stefan-Boltzmann-Gesetz). Der Druck des Photonengases erfüllt , konsistent mit masselosen relativistischen Gasen.
Der **strahlende Energiefluss** , der von einer schwarzen Oberfläche emittiert wird, bezieht sich auf die Energiedichte über , wobei die Stefan-Boltzmann-Konstante ist. Historisch löste Plancks Quantisierungshypothese die ultraviolette Katastrophe in der klassischen Elektrodynamik und legte den Grundstein für die Quantentheorie. Die kosmische Mikrowellenhintergrundstrahlung (CMB), beobachtet bei , liefert eine kosmologische Validierung der Schwarzkörperstrahlungs-Theorie.
3.5.3 Gittervibrationen
In Festkörpern werden Gittervibrationen als Phononen mit linearer Dispersion quantisiert. Das Debye-Modell approximiert den Kristall als ein isotropes Kontinuum mit einer Grenzfrequenz , die sicherstellt, dass es Moden gibt (für Atome in 3D):
Die innere Energie ist:
wobei die Debye-Temperatur ist. Die Wärmekapazität zeigt unterschiedliche Grenzwerte:
Bei hohen Temperaturen (), (Dulong-Petit-Gesetz).
Bei niedrigen Temperaturen (), aufgrund von Phononenmoden, die sich wie masselose Bosonen verhalten.
In Metallen kombiniert die Tieftemperatur-Spezifische Wärme elektronische und phononische Beiträge: , wobei die Fermi-Oberfläche sondiert und phononische Eigenschaften widerspiegelt. Das Debye-Modell ist erfolgreich, indem es die endliche Modendichte und die lineare Dispersion akustischer Phononen erfasst, während Einsteins Modell (Einfrequenz-Oszillatoren) bei niedriger aufgrund seiner exponentiellen Unterdrückung von versagt.